Апатико абулический синдром по мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Патогенез
- Симптомы
- Возможные осложнения
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Профилактика
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Апаллический синдром.
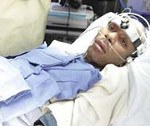
Апаллический синдром
Описание
Апаллический синдром. Клинический симптомокомплекс, включающий отсутствие признаков осознанности при наличии открывания глаз, чередования сон/бодрствования. По мнению неврологов, связан с обширным поражением коры мозга. Может являться переходным состоянием при выходе из комы. Диагностируется исключительно клинически с помощью шкал для оценки уровня сознания. Инструментальные обследования (ЭЭГ, МРТ, МСКТ, ПЭТ, УЗДГ) являются вспомогательными, позволяют установить причинную патологию. Лечение направлено на стимуляцию восстановления, поддержание жизненно важных функций, предупреждение осложнений, уменьшение гидроцефалии.
Дополнительные факты
Новая кора (неокортекс), состоящая из 6 слоёв нейронов, покрывает поверхность полушарий головного мозга и имеет анатомическое название «паллиум» — плащ. Соответственно, апаллический означает отсутствие паллиума, его функциональное «выключение». Термин «апаллический синдром» предложен немецким врачом Кретчмером в 1940, используется в Германии, странах СНГ. В англоязычных источниках по неврологии употребляется введённый в 1972 году термин «вегетативное состояние». Апаллический синдром (АС) встречается у 25-100 пациентов на 1 млн. Человек в популяции, во многих странах отмечается тенденция к увеличению числа случаев. АС наблюдается у лиц обоих полов различных возрастных групп – от грудных младенцев до глубоких стариков.
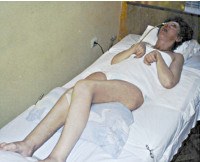
Апаллический синдром
Причины
В основе АС лежит тотальное или субтотальное поражение неокортекса при сохранении функции ствола мозга. Этиологическими факторами выступают:
• Черепно-мозговая травма. Травматический генез имеет большинство случаев АС молодого возраста.
• Церебральная гипоксия. Отмечается при отравлении угарным газом, асфиксии, тяжёлой артериальной гипотонии, остановке сердца вследствие сердечных заболеваний, в ходе оперативных вмешательств.
• Нейроинфекции. Апаллический синдром может становиться исходом острых инфекционных процессов с обширным поражением мозговой коры, возникать на заключительной стадии медленных инфекций ЦНС (лейкоэнцефалита Шильдера, прогрессирующего краснушного панэнцефалита).
• Опухоли головного мозга. В отдельных случаях обширные опухолевые процессы приводят к АС, обусловленному гипоксией, нейротоксикозом, отёком головного мозга.
• Прогрессирующие дегенеративные заболевания. Являются основной причиной АС у пациентов пожилого возраста. Синдром наблюдается на поздней стадии болезни Альцгеймера, Крейтцфельдта-Якоба, Пика, сосудистой деменции, алкогольной энцефалопатии.
• Острые дисметаболические состояния. Уремическая, гипогликемическая, печёночная кома могут переходить в апаллическое состояние.
• Аномалии головного мозга. Микроцефалия, тяжёлая корковая дисплазия. Обуславливают апаллический синдром у детей младшего возраста.
Патогенез
Морфологическая картина коркового поражения при АС неспецифична, различается у разных пациентов. Гипоксические повреждения сопровождаются некрозом, травматические — диффузным аксональным повреждением, дегенеративные — атрофией. Вариабельность объёма и характера поражения, отсутствие в ряде случаев значительных морфологических изменений, свидетельствует о функциональных нарушениях, приводящих к «выключению» коры. Патофизиологические основы этих процессов находятся в стадии изучения.
Переход из комы в апаллическое состояние характеризуется восстановлением функции ретикулярной формации, подкорковых структур. Возобновление связей подкорки и коры приводит к дальнейшему восстановлению сознания. Если корковые связи не восстанавливаются, не формируются вновь или формируются неправильно, возникает хроническое вегетативное состояние — апаллический синдром. В исходе прогрессирующей корковой дегенерации АС развивается вследствие массовой утраты связей, обеспечивающих взаимодействие нейронов внутри коры и с нижележащими структурами.
Симптомы
Основу клинической картины составляет парадокс: наличие видимых признаков сознания при отсутствии объективных критериев осознания пациентом себя и окружающего мира. Больной открывает глаза, двигает ими в состоянии бодрствования, реагирует на болевые раздражения, проходит циклы «сон-бодрствование», что формирует впечатление осознанности. Однако не наблюдается никаких признаков осознанной деятельности, целенаправленной активности. Движения спонтанны, эмоциональные и осознанные реакции отсутствуют. Перемещение глаз хаотично, реакция слежения не наблюдается. Чередование сна и состояния бодрствования не зависит от времени суток.
Лицо пациента маскообразное, без мимики. Жевание и глотание замедлены, могут отмечаться жевательные движения, моргание, зевота. В ответ на болевые стимулы возникает нецеленаправленная двигательная реакция, сопровождающаяся учащением ЧСС и дыхания, расширением зрачков. Функция тазовых органов не контролируется. Возможны эпилептические пароксизмы. Типично повышение мышечного тонуса: кисти сжаты, стопы в состоянии подошвенного сгибания, конечности согнуты и приведены. Функционирование гипоталамуса и мозгового ствола обеспечивает поддержание необходимой гемодинамики, дыхательной деятельности, вегетативной функции. В фазу бодрствования характерно преобладание симпатической нервной системы (учащение ЧСС, АД, возможен озноб), в состоянии сна — парасимпатической (снижение АД, ЧСС, повышенное потоотделение).
Озноб. Судороги.
Возможные осложнения
Вследствие постоянного спастического состояния конечностей развиваются контрактуры суставов. Длительное нахождение пациента в постели с резким ограничением двигательной активности способствует возникновению пролежней, застойной пневмонии. Возможно присоединение вторичной инфекции мочевыводящих путей с развитием пиелонефрита. Апаллический синдром может привести к окончательному угасанию всех мозговых функций с летальным исходом. Смертельно опасными для пациента могут стать инфекционные осложнения, переходящие в сепсис. Правильный уход, питание, поддерживающая терапия способны отсрочить появление осложнений, увеличить продолжительность жизни больного.
Диагностика
Из-за отсутствия чётких критериев сознания и осознанности диагностировать апаллический синдром непросто. Согласно общепринятым правилам, у грудных детей диагноз устанавливается после трехмесячного возраста, поскольку ранее нельзя достоверно дифференцировать осознанное и рефлекторное поведение. В постановке диагноза участвуют неврологи, анестезиологи-реаниматологи, нейрофизиологи, при необходимости — нейрохирурги. Проводятся следующие диагностические мероприятия:
• Неврологический осмотр. Контакт с пациентом полностью отсутствует. Отмечается спонтанное движение глазных яблок, нецеленаправленная реакция на болевые стимулы. Сохранены краниальные и спинальные рефлексы, вегетативная функция. Наблюдается гипертонус мышц конечностей с признаками пластичности, повышение сухожильных рефлексов, наличие симметричных патологических рефлексов.
• Электроэнцефалография. Характерна низковолновая ЭЭГ, дельта- или тета-ритм. У 10-20% больных отмечается альфа- или бета-ритм. Восстановление сопровождается появлением отчётливого альфа-ритма.
• МРТ головного мозга. МРТ и другие способы нейровизуализации (КТ, МСКТ) не выявляют специфических морфологических изменений. Картина соответствует основному заболеванию: определяются очаги некроза, гематомы, опухоли, внутримозговые дегенеративные процессы, отёк мозгового вещества. У 75% больных отмечается гидроцефалия. Большинство случаев АС сопровождаются атрофическими изменениями коры, однако подобная атрофия наблюдается у находящихся в сознании пациентов с деменцией.
• ПЭТ. КТ головного мозга. Позволяет диагностировать снижение коркового метаболизма на 40-50%. Восстановление протекает с активацией обмена веществ, регистрируемой на ПЭТ.
• Транскраниальная УЗДГ. Направлена на оценку церебральной гемодинамики. Имеет вспомогательное значение, используется преимущественно при решении вопроса о целесообразности шунтирующей операции у пациентов с гидроцефалией.
Необходимо дифференцировать апаллический синдром от комы, состояния минимального сознания, сопора.
Дифференциальная диагностика
Дифференцировка осуществляется при помощи клинических шкал. Инструментальные методы не могут точно указать уровень сознания больного, позволяют установить характер поражения коры, судить об уровне метаболизма церебральных тканей.
Лечение
Терапия направлена на жизнеобеспечение пациента, предотвращение осложнений и восстановление сознания. Единый стандарт ведения больных отсутствует. Лечение проводится длительно, иногда месяцами. Применяются консервативные и хирургические методы:
Консервативная терапия:
• Стимуляция восстановительных процессов. Медикаментозная составляющая включает мощную ноотропную, витаминную, сосудистую терапию, эндолюмбальное введение кислорода. Параллельно проводится регулярная сенсорная стимуляция с использованием всего спектра стимулов: слуховых, тактильных, зрительных, обонятельных.
• Искусственное питание. Осуществляется через гастростому, поскольку зондовое питание зачастую сопровождается осложнениями: аспирацией пищи в дыхательные пути, гастроэзофагеальным рефлюксом, изъязвлениями слизистой в местах соприкосновения с зондом.
• Профилактика осложнений. С целью уменьшения спастичности и профилактики контрактур назначают миорелаксанты. Лучшим предупреждением пролежней и гиповентиляционной пневмонии является адекватный уход, включающий смену белья, перемену позы, высаживание при помощи специальных ортопедических приспособлений, пассивную лечебную физкультуру, массаж. Целесообразно приобщение к уходу родственников больного.
Хирургические методы:
• Шунтирующие операции. Показаны при выраженной гидроцефалии. Наиболее распространено люмбоперитонеальное и вентрикулоперитонеальное шунтирование.
• Глубинная электростимуляции мозга. Проводится путём стереотаксического введения микроэлектродов, посредством которых осуществляется стимуляция активирующих систем ствола.
• Нейротрансплантация. Является новой экспериментальной методикой лечения АС. Активирует регенерацию церебральных тканей, обеспечивает материал для реконструкции повреждённых участков. Введение эмбриональных нервных клеток производится интравентрикулярно (в мозговые желудочки), интрацеребрально (в кору или повреждённые глубинные участки мозга).
Прогноз
Исход зависит от характера мозгового поражения, обусловившего апаллический синдром, возраста больного, продолжительности комы, наличия судорожных приступов. Остро развившийся АС может завершиться восстановлением сознания, но в большинстве случаев пациенты остаются инвалидами вследствие выраженного психоорганического синдрома. В случае прогрессирующей церебральной дегенерации апаллический синдром является терминальной стадией и оканчивается летально.
Профилактика
Профилактика возникновения АС заключается в предупреждении травм, нейроинфекций, интоксикаций, своевременном лечении сердечно-сосудистой патологии.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Некоммерческое Партнерство «Рукописные памятники Древней Руси» создано при участии Института русского языка им. В. В. Виноградова РАН и издательства «Языки славянских культур». Научное руководство деятельностью Партнерства осуществляет Научный совет, возглавляемый чл.-корр. РАН, директором Института русского языка РАН А. М. Молдованом. В Научный совет входят: А. А. Гиппиус, В. М. Живов (зам. председателя Научного совета), А. А. Зализняк, А. А. Пичхадзе, Н. Н. Покровский, А. А. Турилов, В. Л. Янин.
Главная наша цель – собрать как можно более обширный электронный архив древнерусских материалов, хранящихся в отечественных и зарубежных музеях (архивах, библиотеках, хранилищах) и представить его в открытом доступе в интернете с соблюдением и защитой всех юридических прав музеев-хранителей рукописей. Важность этой задачи обусловлена тем, что к настоящему времени опубликована лишь незначительная часть (менее 0.1 процента) рукописных памятников, находящихся во многих хранилищах Москвы, Петербурга, Великого Новгорода, Костромы, Твери, Ярославля, Казани, Саратова, и других городов. Между тем собрание древне- и старорусских рукописей (XI–XVII веков), хранящихся в российских библиотеках и архивах, чрезвычайно велико и насчитывает более 100 000 единиц хранения. При отсутствии полноценных и качественных копий мы находимся под постоянной угрозой утраты этого наследия.
Реализация поставленной цели будет осуществляться путем предоставления на нашем сайте персональной экспозиционной площадки музеям, архивам, издательствам, научно-исследовательским коллективам для размещения в открытом доступе электронных копий имеющихся у них рукописных материалов (вместе с необходимой сопутствующей информацией: права на ее публикацию, адреса и телефоны для научных и деловых контактов и пр. Мы также берем на себя техническое обслуживание этой экспозиции, включающее первичную обработку и последующее размещение отсканированной рукописи, обновление и дополнение экспозиционных материалов и др. При этом все права музея, архива (другого правообладателя) на предоставленные материалы полностью сохраняются, а рукописные материалы размещается в виде, не допускающем их несанкционированное издание. Примером может служить наше сотрудничество с Государственной Третьяковской Галереей. В настоящее время нашем сайте размещается электронная копия издания «Типографский Устав (устав с кондакарем)» а трех томах, осуществленного под научной редакцией Б. А. Успенского издательством «Языки славянских культур» при содействии Галереи. Это издание посвящено рукописному памятнику конца XI – начала XII вв. Первый том представляет факсимильное воспроизведение рукописи, второй — наборное воспроизведение и словоуказатель, третий содержит посвященные ей научные исследования. Пример сотрудничества с учеными – раздел, посвященный древнерусским берестяным грамотам, разработанный коллективом ученых во главе с академиками В. Л. Яниным и А. А Зализняком. Впервые в полном объеме представлены коллекции берестяных документов Государственного исторического музея и Новгородского государственного объединенного музея-заповедника. Примером сотрудничества с издательствами является совместная работа с издательством «Языки славянских культур», которое предоставило нам свои издания первых томов Полного собрания русских летописей, п также рукописных книг «История иудейской войны Иосифа Флавия» (древнерусский перевод) и «Псалтырь 1683 г. в переводе Аврамия Фирсова».
Наряду с главной задачей — собрать полный электронный архив древнерусских материалов, мы ставим перед собой еще две задачи.
Вторая задача, непосредственно связанная с первой — создание специальных образовательных программ по изучению самых разных аспектов древнерусской истории, литературы и культуры, как светской, так и религиозной (включая богослужение, монастырское устройство, художественные стили и вкусы, культовые фигуры и проч.). Эти программы, ориентированные на разные образовательные уровни обучающихся (от младших школьников до студентов старших курсов и аспирантов университетов) могут стать незаменимым подспорьем в школьном и вузовском образовании на всей территории России, важным источником возрождения отечественного самосознания и духовных традиций.
Наконец, третья наша задача — способствовать расширению исследований по истории, литературе, культуре, языку Древней Руси, путем облегчения доступа отечественным и зарубежным славистам к рукописным источникам и посвященным им научным работам. Это будет способствовать повышению интереса к русской истории и культуре, росту ее влияния и авторитета в мире.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Апера.
Синдром Апера
Описание
Синдром Апера. Генетическое заболевание, характеризующееся нарушениями процессов окостенения черепа и связанными с этим вторичными расстройствами, а также многочисленными пороками развития скелета и конечностей. Симптомами этого состояния являются карликовый рост, башенная форма черепа, расширенная переносица, незаращение твердого нёба, синдактилии на руках и ногах. Диагностика синдрома Апера производится по характерной клинической картине патологии, на основании рентгенологических данных и молекулярно-генетических исследований. Специфического лечения заболевания не существует, применяют поддерживающую терапию, проводят хирургические вмешательства паллиативного характера.
Дополнительные факты
Синдром Апера (акроцефалосиндактилия 1 типа) – генетическая патология, обусловленная нарушением образования некоторых видов соединительной ткани, главным образом костной. Впервые данное состояние было описано в 1906 году французским педиатром Э. Апером, дальнейшие исследования подтвердили генетическую природу этого заболевания. Этиология и молекулярно-генетические механизмы развития синдрома Апера были определены значительно позднее – лишь в 1995 году. Данная патология может наследоваться по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев ее причиной являются спонтанные мутации в половых клетках родителей (так называемые герминативные мутации).
Синдром Апера с одинаковой частотой поражает как мальчиков, так и девочек, его встречаемость составляет в среднем 1 случай на 160 000-200 000 новорожденных. Врачи-генетики в настоящее время относят синдром Апера к особой группе наследственных заболеваний – акроцефалосиндактилиям, характеризующиеся одновременным поражением костей черепа и конечностей. Особенностью этой патологии является важность ее как ранней диагностики, поскольку паллиативные мероприятия в раннем возрасте могут в значительной степени влиять на дальнейшее интеллектуальное развитие больного.
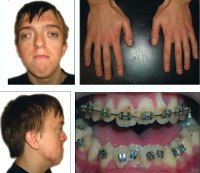
Синдром Апера
Причины
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной. Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями. Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R. Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния. Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности – к преждевременному зарастанию швов и остановке нормального роста черепной коробки. Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов. Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR. Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей – различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Симптомы
Некоторые проявления синдрома Апера заметны с самого рождения – например, синдактилия, которая может быть полной или в виде перепонок. Как правило, срастаются 2, 3 и 4 пальцы на кистях, иногда аналогичный порок возникает и на пальцах ног. Среди неонатологов симптом иногда носит название «среднего пальца» – в тяжелых случаях эти три пальца прочно срастаются между собой и имеют один общий ноготь. Другим постоянным симптомом синдрома Апера, обнаруживающимся сразу после рождения или в первые месяцы жизни, является раннее развитие синостоза костей черепа. Чаще всего происходит срастание венечного или стреловидного шва, что по мере роста головного мозга приводит к деформации черепа по типу «башенной». Из-за черепного синостоза у больных синдромом Апера наблюдается хроническое повышение внутричерепного давления, становящееся причиной задержки умственного развития, головных болей, тошноты и рвоты.
Помимо деформации черепа о наличии синдрома Апера свидетельствует характерный внешний вид больных. У них обычно обнаруживается плоский или выпуклый лоб, гипертелоризм и экзофтальм, может развиваться косоглазие. Деформации затрагивают и кости лицевого черепа – переносица расширена, челюсти нередко недоразвиты, наблюдается нарушение прикуса. Из других симптомов синдрома Апера иногда регистрируются нарушения дыхания (из-за недоразвития верхней челюсти, сужения хоан или трахеи), незаращение твердого нёба, врожденные пороки сердца, аномалии развития позвонков, почек, прямой кишки.
Рвота. Тошнота.
Диагностика
Диагностика синдрома Апера производится на основании осмотра и изучения настоящего статуса пациента, рентгенологических исследований, молекулярно-генетических анализов. При осмотре у больного выявляется синдактилия (у лиц старшего возраста могут обнаруживаться следы ее хирургической коррекции), деформация черепа – башенный череп или брахикефалия, характерный внешний вид лица. С возрастом у больных синдромом Апера могут нарастать признаки нарушения дыхания, при ЭхоКГ нередко определяются пороки сердца и сосудов – стеноз легочного ствола или аорты, дефекты межжелудочковой перегородки. Иногда на этом фоне выявляются признаки сердечной недостаточности. Также возможно наличие иных пороков развития – аномалий позвонков, глухоты, слепоты (из-за катаракты, пигментного ретинита, атрофии зрительных нервов), патологий почек и поджелудочной железы. Из-за столь широкого спектра возможных нарушений больные синдромом Апера нуждаются в тщательном и всестороннем медицинском обследовании.
Рентгенологическими методиками уже у маленьких детей можно обнаружить синостоз костей черепа в области венечного или стреловидного шва. В дальнейшем при помощи рентгенографии можно определить характерную для синдрома Апера деформацию черепной коробки, пороки развития костей лицевого черепа, аномалии позвонков и другие нарушения. Наиболее достоверным диагностическим методом при этом состоянии является молекулярно-генетический анализ. Как правило, для выявления синдрома Апера производят секвенирование 7 экзона гена FGFR2, иногда используют менее затратные техники, ориентированные только на поиск наиболее распространенных мутаций (S252W и P253R), приводящих к этому заболеванию. Подобные методики более дешевые и быстрые в выполнении, обладают точностью на уровне 95%, возможно их использование в качестве пренатальной диагностики этого состояния. Подобный анализ особенно актуален, если посредством профилактических УЗИ у плода выявляются нарушения, предположительно связанные с синдромом Апера – пороки развития черепа, сердца, верхних или нижних конечностей.
Лечение
Специфического лечения синдрома Апера на сегодняшний день не существует, однако паллиативные и симптоматические мероприятия могут значительно облегчить состояние больного и улучшить качество его жизни. Особенно важно как можно раньше диагностировать это заболевание по той причине, что своевременная хирургическая коррекция черепного синостоза позволит избежать значительного роста внутричерепного давления. По многочисленным данным, после таких операций, произведенных в раннем детстве, признаки умственной неполноценности у больных синдромом Апера были выражены значительно слабее, иногда сохранялся нормальный интеллект. Поэтому борьба с внутричерепной гипертензией играет центральную роль в паллиативном лечении этого состояния. Если же у пациентов имеется умственная отсталость, то ее выраженность снижается путем психокоррекционной работы.
Другой часто выполняемой паллиативной хирургической операцией при синдроме Апера является вмешательство для разделения сросшихся пальцев на руках и ногах. Это относительно несложная процедура при перепончатом типе сращения, однако при более тяжелых формах порока операция значительно усложняется. При синдроме Апера также может потребоваться помощь хирургов в случае пороков сердца, сужения хоан или трахеи, нарушения формирования прямой кишки и других проявлений этого генетического заболевания. Больные нуждаются в регулярных медицинских обследованиях у специалистов различного профиля.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа. Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком. Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник