Врожденный нефротический синдром финский тип

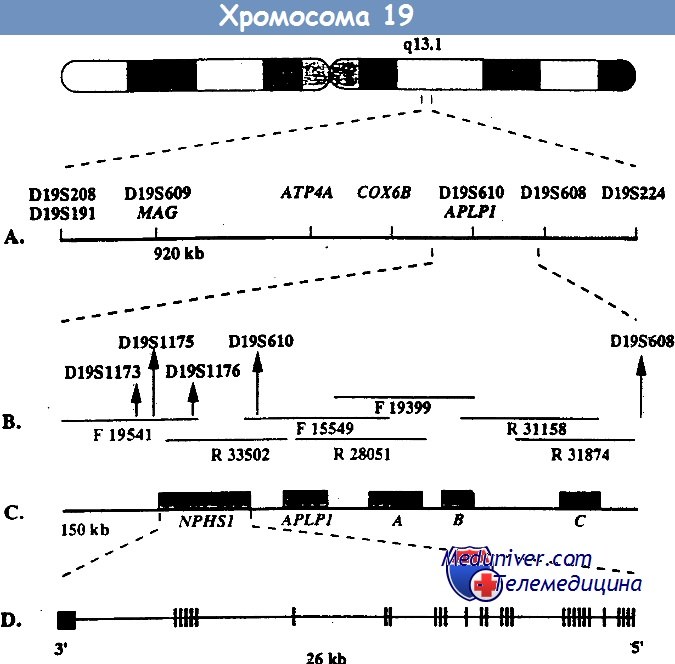
Врожденный нефротический синдром финского типа — клиника, диагностикаВрожденный нефротический синдром финского типа — заболевание наследуется по аутосомно-рецессивному типу и является основной причиной высокой протеинурии у детей первого месяца жизни. Хотя наибольшая распространенность этого заболевания отмечается в Финляндии (1,2 случая на 10 000 беременностей), описано много случаев заболевания у детей других национальностей. При этом заболевании протеинурия возникает еще внутриутробно, что проявляется повышенным уровнем а-фетопротеина в околоплодных водах. Уже на первой неделе жизни часто возникают отеки. Истощение, тяжелые инфекции и тромбозы обусловливают тяжесть заболевания и высокую смертность, ранее больные погибали на первом году жизни. Сегодня при интенсивном лечении больные могут дожить до того момента, когда им можно провести трансплантацию почки. Выживаемость как трансплантата, так и больных очень высокая. Локус, мутация в котором обусловливает данное заболевание, был найден с помощью позиционного клонирования на длинном плече 19-й хромосомы (19q13.1) и в финских, и в других семьях. При определении нуклеотидной последовательности этого локуса был найден ранее неизвестный ген NPHS1, который избирательно экспрессируется в подоцитах. Продукт этого гена получил название нефрин. Он относится к молекулам адгезии из суперсемейства иммуноглобулинов. Нефрин локализован в области щелевых диафрагм — видоизмененных плотных контактов между отростками ножек подоцитов. У больных с мутацией гена NPHS1 нет отростков ножек подоцитов и щелевых диафрагм. Это позволяет думать, что именно нефрин является важнейшим компонентом щелевых диафрагм, предотвращающих выход белка из сосудов клубочка. Среди всех мутаций гена NPHS1 у финнов преобладают две: Fin-major и Fin-minor. Они присутствуют более чем у 90% больных. Мутация Fin-major вызвана делецией двух пар нуклеотидов во 2-м экзоне, который кодирует терминирующий кодон, она встречается примерно у 80% больных финнов. Мутация Fin-minor — нонсенс-мутация в 26-м экзоне, она встречается примерно у 17% больных финнов. У больных других национальностей встречаются различные мутации по типу делеций, вставок, нонсенс- и миссенс-мутаций, а также мутации, нарушающие сплайсинг. Врожденный нефротический синдром финского типа — основная, но не единственная причина нефротического синдрома на первом месяце жизни.
— Также рекомендуем «Синдром Дени-Дрэша — клиника, диагностика» Оглавление темы «Наследственные болезни почек»:
|
Источник
Вторичный и врожденный нефротические синдромы у детей. Диагностика
Нефротический синдром может быть также вторичным проявлением многих поражений почечных клубочков: мембранозная нефропатия, мезангиокапиллярный, постинфекционный и волчаночный гломерулонефрит, геморрагический васкулит. Вторичный нефротический синдром следует подозревать у больных старше 8 лет при наличии артериальной гипертонии, гематурии, нарушения функции почек, внепочечных симптомов (сыпь, артралгия и др.) или низкого уровня комплемента в крови.
В некоторых регионах мира ведущими причинами нефротического синдрома являются малярия и шистосомоз. Вызывают нефротический синдром вирусы гепатита В и С, филярии, возбудитель проказы и ВИЧ.
Нефротический синдром сопутствует злокачественным опухолям, особенно у взрослых. У больных с солидными опухолями (например раком легкого, желудка или кишечника) почечная патология часто напоминает мембранозную нефропатию. По видимому, в этих случаях в почках откладываются комплексы опухолевых антигенов со специфическими антителами. При лимфомах, особенно лимфоме Ходжкина, поражение почек чаще всего напоминает болезнь минимальных изменений. Предполагается, что продуцируемый лимфомой лимфокин увеличивает проницаемость стенки клубочковых капилляров. Нефротический синдром возможен и до обнаружения опухоли, но исчезает при ее регрecce и возобновляется при рецидиве.
К развитию нефротического синдрома приводит также многочисленные лекарственные средства и химические вещества. Гистологическая картина почек в этих случаях может напоминать мембранозную нефропатию (пенициллин, каптоприл, препараты золота, НПВС, соединения ртути), болезнь минимальных изменений (пробенецид, этосуксимид, метимазол, литий) или мезангиокапиллярный гломерулонефрит (прокаинамид, хлорпропамид, фенитоин, триметадион, параметадион).
Врожденный нефротический синдром у детей
При развитии нефротического синдрома у детей первых 3 мес. жизни он считается врожденным. Чаще всего наблюдается врожденный нефротический синдром финского типа — аутосомно-рецессивное заболевание, наиболее распространенное среди потомков выходцев из Скандинавии (частота 1:8000). В его основе лежит мутация расположенного на хромосоме 19 гена NPHS1, который кодирует белок нефрин (важнейший компонент фильтрационных щелей между ножками подоцитов). Считается, что нефрин определяет нормальную функцию фильтрационного барьера в почечных клубочках. Гистологически синдром характеризуется в основном расширением проксимальных канальцев, пролиферацией клеток мезангия и склерозом клубочков.
У грудных детей обнаруживаются массивная протеинурия (которую можно определить и внутриутробно по повышению уровня а-фетопротеина) и отеки. Плацента увеличена. Больные часто рождаются недоношенными с респираторным дистрессом и расхождением швов черепа. Отеки сохраняются, часто возникают инфекционные заболевания, и к 5-летнему возрасту больные обычно погибают от почечной недостаточности. Кортикостероиды и иммуносупрессивные средства неэффективны.
Уменьшить протеинурию и улучшить состояние больных можно с помощью ингибиторов АПФ, индометацина и односторонней нефрэктомии. Однако в этих случаях чаще выполняют двустороннюю нефрэктомию с последующим постоянным диализом и активными диетическими мерами. В конце концов требуется трансплантация почки. В семьях, где имелись или имеются больные с врожденным нефротическим синдромом финского типа, для пренатальной диагностики определяют уровень а-фетопротеина в амниотической жидкости. Диагноз можно подтвердить данными анализа ДНК.
Другие причины врожденного нефротического синдрома включают врожденные инфекции, такие как сифилис, токсоплазмоз, краснуха и ЦМВ-инфекция, а также ВИЧ и вирус гепатита В. Поражение почек в этих случаях выражено слабее, чем при врожденном нефротическом синдроме финского типа, и излечение основного заболевания смягчает или устраняет почечную патологию.
У небольшого числа больных с врожденным нефротическим синдромом наблюдается диффузный мезангиальный склероз с прогрессирующим склерозом мезангия клубочков и быстрым ухудшением функции почек. Терминальная стадия почечной недостаточности развивается за несколько месяцев или лет. Диффузный мезангиальный склероз может быть как отдельным заболеванием, так и проявлением синдрома Дени-Дрэша (сочетание нефробластомы с мужским псевдогермафродитизмом). В основе этого синдрома лежит мутация гена опухоли Вильмса (WT1) на хромосоме 11.
— Также рекомендуем «Функция канальцев почек. Канальцевая фильтрация»
Оглавление темы «Заболевания почек у детей»:
- Нефротический синдром у детей. Причины и диагностика
- Идиопатический нефротический синдром у детей. Диагностика
- Лечение нефротического синдрома у детей. Гормоны
- Осложнения нефротического синдрома у детей. Прогноз
- Вторичный и врожденный нефротические синдромы у детей. Диагностика
- Функция канальцев почек. Канальцевая фильтрация
- Почечный канальцевый ацидоз. Проксимальноканальцевый
- Цистиноз у детей. Синдромы Лоу и Фанкони
- Дистальноканальцевый ацидоз у детей. Клиника
- Гиперкалиемический некроз почечных канальцев у детей. Диагностика и лечение
Источник
Врожденный нефротический синдром – НС, развившийся у детей с момента рождения или в первые 3 месяца жизни. У детей он гетерогенен, выделяют первичные и вторичные формы врожденного НС. В большинстве случаев доминируют первичные формы, которые представляют собой гетерогенную группу заболеваний с различной этиологией и прогнозом: ВНС финского типа, диффузный мезангиальный склероз, изолированный или в комплексе синдрома и липоидный нефроз. Вторичные формы ВНС чаще развиваются на фоне инфекционных заболеваний, таких как цитомегалия, сифилис, токсоплазмоз, ВИЧ.
А. Первичные формы:
— Врожденный НС финского типа
— Врожденный НС французкого типа
— Другие НС (с минимальными изменениями, ФСГС, мембранозный ГН)
— Синдромальные аномалии (синдром Галловей-Моуат, врожденный НС с аномалиями нервной системы и другие синдромы)
В. Вторичные формы:
— На фоне инфекционных заболеваний (врожденный сифилис, токсоплазмоз, краснуха, цитомегалия, малярия)
— При СКВ у матери
— НС ассоциированный с тромбозом почечных вен
Врожденный НС финского типа– наиболее частый вариант врожденного НС. Заболевание чаще встречается в Финляндии – 1:8200 рождений, но может регистрироваться и в других регионах мира у лиц различных национальностей, в России – чаще регистрируется в западной части страны. Это генетическое заболевание, которое наследуется аутосомно-рецессивно. В 1994 году был изолирован ген, локализующийся на 19 хромосоме. Этот ген (NPHS1) кодирует белок – нефрин, который является трансмембранным белком суперсемейства иммуноглобулинов. В почках этот белок выявлен на щелевидной мембране между ножками подоцитов. В финских семьях регистрируют 4 мутации этого гена, наиболее часто регистрируются: Fin-major и Fin-minor мутации. Другие мутации (46 вариантов) регистрируют у лиц нефинской национальности. Девочки и мальчики болеют одинаково часто.
Клиника и диагностика. Большинство детей рождаются преждевременно между 35-й и 38-й неделями беременности. Масса плаценты достигает более 25% от массы тела новорожденного. Соотношение массы плаценты к массе тела ребенка увеличено до 0,43 (при норме 0,18). Во время беременности у женщин с 16-й по 20-ю неделю беременности определяется повышенный уровень α-фетопротеина в амниотической жидкости или в сыворотке крови матери. Заболевание проявляется полным клинико-лабораторным симптомокомплексом гормонорезистентного НС, нередко с микрогематурией. АД нормальное. Массивная протеинурия, представленная в 90% случаев альбуминами начинается еще во внутриутробном периоде. В 25% случаев массивные отеки появляются с рождения, в 90% — на первой неделе. Гипоальбуминемия часто достигает критического уровня (ниже 5 г/л), определяется гиперлипидемия. Прогрессируют нарушения функций почек. ХПН развивается к 4 годам. На УЗИ почки увеличены в размерах с повышенной эхогенностью паренхимы или тотальной гиперэхогенностью при отсутствии четкой кортикомедуллярной дифференцировки. Морфологически на ранних сроках заболевания может отсутствовать патогномоничная гистологическая картина, с 3 месяцев отмечается микрокистозная дилятация канальцев, в основном проксимальных. Сами клубочки могут быть не изменены, либо в них отмечаются пролиферативные процессы. Прогноз неблагоприятный.
Врожденный НС французского типа– симптомокомплекс НС, проявляющийся в первые 3 месяца жизни и характеризующийся морфологически диффузным мезангиальным склерозом. Заболевание имеет аутосомно-рецессивный путь наследования. Девочки и мальчики болеют одинаково часто. Дети рождаются доношенными с нормальным весом при рождении. Масса плаценты нормальная. Уровень α-фетопротеина у матери во время беременности в норме. НС в 67% сочетается с микрогематурией и характеризуется резистентностью и отсутствием эффекта от другой иммуносупрессивной терапии. Артериальная гипертензия определяется у 71% больных. ХПН обычно развивается к 1,5-2 годам.
Лечение.Цель – довести ребенка до возраста, приемлемого для трансплантации, которая является единственным средством излечения. Протокол, предложенный финскими педиатрами в начале 90-х г. включает:
1) Компенсацию гипоальбуминемии (20% альбумин) в сочетании с фуросемидом до уровня альбумина сыворотки 15-20 г/л.
2) Заместительную терапию (витамин Д, тироксин, витамины, кальций).
3) Питание (энтеральное через назогастральный зонд 130 ккал/кг, 4 г/кг/сут белка, жидкость 100-130 мл/кг/сут; 10-14% белок, 40-50% липиды, 40-50% углеводы).
4) Профилактику и лечение тромботических осложнений (курантил, гепарин, низкомолекулярные гепарины).
5) Применение ингибиторов АПФ (капотен).
6) Профилактику и лечение инфекционных осложнений
Такая тактика продолжается, пока ребенок не достигнет массы тела около 7 кг – возраста, в котором проводится бинефрэктомия. В дальнейшем ребенок находится на перитониальном диализе или гемодиализе до достижения параметров, необходимых для проведения трансплантации почки, которую осуществляют после достижения массы тела 9 кг.
Наследственный нефрит
Наследственный нефрит – генетически детерминированное неиммунное нефритоподобное заболевание, проявляющееся гематурией и (или) протеинурией и часто сочетающееся с патологией слуха и реже зрения. Заболевание передается по аутосомно-доминантному типу, сцепленному с Х-хромосомой (80-85%), аутосомно-рецессивному или аутосомно-доминантному типу наследования. Генные мутации приводят к нарушению трехспиральной структуры коллагена (альфа цепи коллагена 4 типа), что вызывает изменение не только базальных мембран почки, но и аналогичных структур уха и глаза. Выделяют 3 варианта наследственного нефрита.
1. Синдром Альпорта, для которого характерно наследственный нефрит с гематурией, тугоухость и поражение глаз. Заболевание наследуется по доминантному, сцепленному с Х-хромосомой, типу наследования. Течение нефрита прогрессирующее с исходом в хроническую почечную недостаточность.
2. Наследственный нефрит без тугоухости, характеризующий прогрессирующим течением с исходом в хроническую почечную недостаточность. Заболевание наследуется по доминантному, сцепленному с Х-хромосомой, типу наследования.
3. Семейная доброкачественная гематурия, которая протекает доброкачественно с благоприятным прогнозом. Заболевание наследуется по аутосомно-доминантному или аутосомно-рецессивному типу наследования. При аутосомно-доминантном типе наследования отмечается тромбоцитопения.
При морфологическом исследовании определяются диспластические, дистрофические, пролиферативные изменения, фокально-сегментарный гломерулосклероз. Прогрессирование поражения ведет к атрофии и дистрофии канальцев, интерстициальному фиброзу. При электронной микроскопии выявляются истончение, расщепление, нарушение структуры базальной мембраны. Клиническая картина разнообразна по развитию, проявлениям и течению. Выделяют 3 стадии течения нефрита: в первую стадию самочувствие ребенка не страдает, отмечается изолированный мочевой синдром, нет нарушения функции почек; вторая стадия характеризуется ухудшением самочувствия, нарастанием изменений в моче и почечной недостаточности тубулярного типа; третья стадия – терминальная — развивается к 20-30 годам, иногда раньше.
Первые признаки поражения почек при синдроме Альпорта обычно выявляют в возрасте от 3 до 10 лет. Обычно их выявляют случайно, в виде изолированного мочевого синдрома. Наиболее частым и первым признаком заболевания является гематурия различной степени выраженности. Но иногда ранним признаком заболевания бывает и протеинурия или, реже, снижение слуха. Обычно эти признаки выявляют в среднем в 6-летнем возрасте.
Гематурия при наследственном нефрите может спонтанно как появляться, так и исчезать. Очень часто она провоцируется острой респираторной вирусной инфекцией. Эритроциты в моче обычно дисморфичны, обычно обнаруживают эритроцитарные цилиндры. Протеинурии может не быть в первые годы, нередко она минимальная и имеет интермиттирующий характер. Редко отмечаются протеинурия более 2 г/сут и развитие нефротического синдрома.
Возможен наследственный нефрит с тромбоцитопенией и лейомиоматозом. Первоначально выявляют лейомиому пищевода (доброкачественная опухоль, исходящая из мышечной оболочки) с преимущественной локализацией в грудной его части. Трахеобронхиальная локализация встречается реже, но она может быть причиной летального исхода вследствие бронхоспазма. Несколько позже появляется лейомиома половых органов. Описаны случаи локализации лейомиом в области клитора, малых и больших половых губ.
У девочек заболевание чаще проявляется рецидивирующей гематурией. У мальчиков клиническое течение заболевания более тяжелое, чем у девочек. Ухудшению состояния способствуют интеркуррентные заболевания, усиление физической нагрузки, инсоляция.
Глухота чаще встречается у мальчиков, чем у девочек, развивается приблизительно к 10 годам. Снижение слуха выявляют у 74% мальчиков и у 5% девочек. Оно имеет неврогенное происхождение, выражено в различной степени, с возрастом прогрессирует от умеренного до полного. На ранних этапах снижение слуха происходит на высоких частотах, распространяясь позднее на более низкие, переходя из звукопроводящей в звуковоспринимающую тугоухость. На ранней стадии заболевания при аудиометрии выявляют невосприимчивость звуков с частотой 6-8 кГц, а впоследствии и более низких частот (4,1-2 кГц). Поражение VIII пары черепномозговых нервов или кортиева органа бывает чаще двусторонним. Ранняя тугоухость косвенно указывает на тяжесть почечного процесса. При гистологическом исследовании внутреннего уха выявляют различные изменения, среди которых чаще всего — потерю нейронов и волосяных клеток, атрофию спиральных связок, дегенерацию stria vascnlaris.
Глазные аномалии проявляются изменением полей зрения, аномалиями хрусталика и роговицы. Для синдрома Альпорта свойственны катаракта, задний лентиконус, задняя полиморфная дистрофия роговицы, псевдоотек сосочков, дистрофия сетчатки, телеангиэктазия сетчатки, нарушение цветового восприятия, колобома, страбизм, нистагм, прогрессирующий двусторонний кератоконус. Нередко выявляют нистагм и миопию. При офтальмологическом исследовании у больных часто выявляется снижение остроты зрения, передний лентиконус, пятна на сетчатке, катаракта, кератоконус.
Микроневрологическая симптоматика встречается у 90% больных с наследственным нефритом. У трети больных отмечаются симптомы вегетативной дисфункции — колебания АД, эмоциональная лабильность, головная боль, гипергидроз ладоней и стоп. Иногда определяются симптомы пирамидной недостаточности (гиперрефлексия и др.), сглаженность носогубных складок, асимметрия сухожильных рефлексов.Нарушения памяти и снижение интеллекта встречаются редко.
Для наследственного нефрита характерны признаки дисэмбриогенеза. На экскреторных урограммах иногда выявляют лоханочную эктазию, удвоенную почку, патологическую подвижность, незавершенный поворот почки.
При наследственном нефрите наблюдаются снижение уровней Т- и В-популяций лимфоцитов, IgA, склонность к повышению концентраций IgM и IgG. Снижена фагоцитарная активность. Снижение общей резистентности организма предрасполагает к пиелонефриту, гнойному отиту, частым простудным заболеваниям.
Функциональное состояние почек сохранено в стадии скрытых клинических проявлений или компенсации. В стадии субкомпенсации превалируют ренальные дисфункции по тубулярному типу с исходом в тотальную ХПН. При наследственном нефрите в биоптатах почек у детей с возрастом увеличиваются соотношение интерстиций/кора и количество склерозированных гломерул, которые являются маркерами рубцевания почек.
В ранние сроки заболевания диагностировать заболевание сложно, поскольку нет патогномоничных симптомов. Диагноз синдрома Альпорта устанавливают на основании обнаружения у ребенка нефропатии с гематурией при наличии в семье больного с аналогичной патологией и сочетании поражения почек с глухотой у самого больного или кого-то из членов семьи. Поэтому для постановки диагноза важно составлять родословную семьи больного.
Согласно данным Clifford et al. (1993), диагностическим критерием служит наличие 3 из 5 признаков, один из которых относится к почкам:1)гематурия или смерть от ХПН в семейном анамнезе; 2) гематурия или нефротический синдром у пациента; 3) изменения гломерулярных базальных мембран (при электронной микроскопии биоптата почки); 4) снижение слуха (по данным аудиограммы); 5) врожденная патология зрения.
Для подтверждения диагноза используют биопсию почек. Для синдрома Альпорта характерны неравномерность контуров гломерулярной базальной мембраны, расслоение или сетеобразность ее плотной пластинки.
Эффективных методов патогенетической терапии наследственного нефрита нет. Лечение предусматривает организацию щадящего режима. Ограничивают физические нагрузки, не проводят профилактические прививки. Диета высококалорийная, сбалансированная, с учетом функционального состояния почек. При отсутствии признаков нарушения функции почек назначают диету с достаточным содержанием белков, жиров и углеводов. Но диета с ограничением белков, липидов, кальция и фосфора отдаляет сроки развития ХПН. Сообщено об успешном применении в комплексном лечении наследственного нефрита ингибиторов ангиотензинпревращающего фермента, которые уменьшают выраженность протеинурии и замедляют прогрессирование заболевания. Используют активаторы обмена, как пиридоксин (по 2-3 мг/кг/сут в 3 приема в течение 2-4 нед), кокарбоксилаза (по 50 мг внутримышечно через 1 сут; 10-15 инъекций), АТФ (по 1 мл внутримышечно через 1 сут; 10-15 инъекций), витамин А (по 1000 ЕД/год жизни в сутки в 1 прием; 10-14 сут), витамин Е (по 1 мг/кг/сут за 1 прием; 10-14 сут). Указанные препараты назначают курсами 2-3 раза в год. Эффективна также фитотерапия. В качестве иммуностимуляторов назначают левамизол (декарис) по 2 мг/кг/сут 2-3 раза в 1 неделю с 4-дневным перерывом. При развитии хронической почечной недостаточности проводится гемодиализ и трансплантация почки. Успех диализа и трансплантации зависит от подбора трансплантата и наличия антител к ГБМ. Антибактериальная, иммуносупрессивная и стероидная терапия показаны в пред- и посттрансплантационный периоды. Коррекцию зрения проводят с помощью очков или контактных линз. Описан положительный опыт имплантации хрусталика и оперативного лечения переднего лентиконуса.
Больные с наследственным нефритом находятся на диспансерном учете в течение всей жизни. Прогностически неблагоприятными критериями течения наследственного нефрита являются: принадлежность к мужскому полу; раннее развитие ХПН у членов семьи; протеинурия (уровень протеина более 1 г/сут); утолщение гломерулярных базальных мембран (при электронной микроскопии); неврит слухового нерва и делеция в гене COL4A5.
Источник