Синдром пфайфера что это такое


Синдром Пфайффера – генетическое заболевание с аутосомно-доминантным механизмом наследования, характеризующееся нарушением формирования костей черепа и конечностей. Симптомами являются деформации черепа (в результате краниосиностоза), костей пальцев рук и ног. Некоторые формы заболевания характеризуются глухотой, нарушением интеллектуального развития, стоматологическими патологиями. Диагностика синдрома Пфайффера производится на основании данных настоящего статуса больного, рентгенологических исследований, молекулярно-генетических анализов. Специфическое лечение отсутствует, применяют паллиативные мероприятия для предотвращения тяжелых осложнений (в том числе и хирургического характера) и симптоматическую терапию.
Общие сведения
Синдром Пфайффера – врожденное генетически обусловленное нарушение формирования черепа и конечностей, которое характеризуется краниосиностозом, деформацией лица, расширением и укорочением пальцев, частыми синдактилиями. Впервые это заболевание было описано в 1964 году немецким педиатром Рудольфом Пфайффером, в дальнейшем было выявлено несколько клинических форм патологии. По данным современной генетики, синдром Пфайффера является аутосомно-доминантным состоянием с полной пенетрантностью, половое распределение больных не имеет особенностей – с одинаковой частотой болеют как мальчики, так и девочки. Встречаемость точно не определена, по некоторым данным она составляет примерно 1:100 000 новорожденных. Каких-либо национальных или расовых особенностей в распределении этого заболевания также не выявлено. В отношении синдрома Пфайффера крайне важна ранняя диагностика, так как от времени начала паллиативного лечения во многом зависит прогноз заболевания и качество дальнейшей жизни больного.

Синдром Пфайффера
Причины синдрома Пфайффера
Несмотря на то, что синдром Пфайффера является наследственным заболеванием с аутосомно-доминантным механизмом передачи, большинство случаев этой патологии являются следствием спонтанных герминативных мутаций в половых клетках родителей. При этом за развитие состояния отвечают дефекты двух генов, кодируемые ими белки обладают сходными функциями. Первый ген – FGFR1, расположен на 8-й хромосоме и кодирует последовательность белка-рецептора к фактору роста фибробластов-1. Наиболее частым типом генетического дефекта при этом становится миссенс-мутация 252Pro-Arg в экзоне 7а. Другим геном, мутации которого приводят к развитию синдрома Пфайффера, является FGFR2, расположенный на 10-й хромосоме – продуктом его экспрессии также является рецептор к фактору роста фибробластов (РФРФ-2). В этом гене выявлено более 26 различных вариантов мутаций, способных приводить к различным формам синдрома Пфайффера.
По причине того, что вышеуказанные белки обладают очень похожими функциями, патогенез синдрома Пфайффера при дефектах гена FGFR1 и FGFR2 тоже весьма схож. В результате миссенс-мутаций изменяется структура экспрессируемого белка, из-за чего полученный рецептор становится неспособен полноценно связываться с веществом-стимулятором (фактором роста фибробластов) и передавать информацию внутрь клетки. Так как фибробласты активно участвуют в процессах остеогенеза в эмбриональный период и в первые годы жизни ребенка, такое изменение рецепторов ведет к разнообразным костным патологиям.
Доказательством этого является тот факт, что дефекты гена FGFR1, помимо синдрома Пфайффера, приводят к тригоноцефалии 1-го типа и ряду других нарушений, а FGFR2 – к неспецифическому краниосиностозу, синдрому Крузона и иным похожим патологиям. При синдроме Пфайффера происходит преждевременное сращение костей черепа, что становится причиной деформации головы и лица, повышения внутричерепного давления – это, в свою очередь, способно вызывать ряд вторичных нарушений от нейросенсорных расстройств до глубокой умственной отсталости.
Классификация и симптомы синдрома Пфайффера
На сегодняшний день выделяют как минимум три клинические формы синдрома Пфайффера, которые различаются между собой выраженностью нарушений, течением и прогнозом заболевания. Причина такого различия лежит в молекулярно-генетических механизмах развития патологии – 1-й тип обусловлен единственной мутацией гена FGFR1 (252Pro-Arg) и некоторыми нарушениями структуры FGFR2, тогда как 2-й и 3-й типы вызваны различными вариантами дефектов только гена FGFR2. Более тонкие особенности патогенеза каждой формы заболевания в настоящее время находятся в стадии изучения. Благодаря характерной клинической картине, специалисты могут определить разновидность синдрома Пфайффера, основываясь только на симптомах патологии. Молекулярно-генетический анализ во многих случаях необходим лишь для окончательного подтверждения диагноза.
Синдром Пфайффера 1-го типа является наиболее распространенным и благоприятным в прогностическом отношении вариантом заболевания. При этом состоянии уже с самого рождения ребенка отмечаются аномалии развития лица – гипертелоризм, недоразвитие верхнечелюстных костей, расширенные плоские переносица и спинка носа. Иногда могут отмечаться расщепление твердого нёба и экзофтальм. При этой форме синдрома Пфайффера характерные изменения формы черепа сводятся к увеличению его высоты и длины, очень редко отмечается асимметрия. В дальнейшем могут возникать аномалии зубов – искривление зубного ряда, гипоплазия, склонность к кариесу. Деформации костей конечностей сводятся к расширению фаланг первых пальцев на руках и ногах, может отмечаться частичная синдактилия.
Синдром Пфайффера 2-го типа – более тяжелая форма заболевания, проявляющаяся значительной выраженностью сращения костей черепа между собой и рядом других аномалий. Голова из-за краниосиностоза приобретает характерную форму «трилистника» или «листка клевера», наблюдается значительная гипоплазия средней трети лица. По причине грубого нарушения формирования черепа данный тип синдрома Пфайффера характеризуется выраженной умственной отсталостью и рядом неврологических нарушений. Пороки развития конечностей сводятся к расширению первых пальцев, синдактилии, анкилозу локтевых суставов. Кроме того, синдром Пфайффера 2-го типа часто сопровождается различными пороками развития внутренних органов. Все вышеперечисленные нарушения могут стать причиной летального исхода в раннем возрасте.
Синдром Пфайффера 3-го типа во многом схож по своим проявлениям с предыдущим вариантом заболевания, но краниосиностоз при этом не приводит к формированию деформации черепа в виде «листка клевера». У больных отмечается увеличение головы в высоту, пороки развития конечностей (синдактилии, анкилозы суставов), экзофтальм, неврологические нарушения. Для этого варианта синдрома Пфайффера характерно раннее развитие зубов, нередко младенцы уже рождаются с ними (натальные зубы). Также возможны пороки внутренних органов и ранняя смерть из-за совокупности нарушений.
Такое различие в клинической картине и прогнозе различных вариантов синдрома Пфайффера накладывает свой отпечаток на наследование этого заболевания. Так как 1-й тип, особенно в случае своевременного выявления и правильного лечения, характеризуется сохранением нормального уровня интеллекта, фертильности и выживаемости больных, возможна аутосомно-доминантная передача патологии потомству. Более тяжелые 2-й и 3-й типы чаще всего приводят к ранней смерти или глубокой инвалидизации больных, эти варианты синдрома Пфайффера возникают только в результате спонтанных мутаций.
Диагностика и лечение синдрома Пфайффера
Своевременная диагностика синдрома Пфайффера крайне важна, так как она позволяет вовремя составить схему паллиативного лечения, тем самым избежать осложнений и значительно улучшить качество жизни больного. Но это справедливо лишь в отношении 1-го типа заболевания, так как при других вариантах патологии комплекс пороков развития настолько тяжел, что практически не оставляет шансов на сохранение интеллекта, а в дальнейшем – жизни больных. Для выявления синдрома Пфайффера используют ультразвуковые методики (в том числе и в качестве пренатальной диагностики), рентгенографию, молекулярно-генетические анализы. На профилактических УЗИ во время беременности при наличии этого заболевания у ребенка во 2-3 триместре можно выявить нарушение формирования черепа, синдактилию, пороки развития внутренних органов.
Рентгенологические методы редко используют при ранней диагностике синдрома Пфайффера у детей – в основном, производят исследование черепа, которое обнаруживает наличие краниосиностоза различной степени выраженности, гипоплазию верхнечелюстных костей, изменение формы глазниц. Также на рентгене можно определить изменение формы костей больших пальцев рук и ног. Молекулярно-генетическая диагностика производится врачом-генетиком – в этом случае при подозрении на наличие синдрома Пфайффера 1-го типа производят поиск точечной мутации 252Pro-Arg методом прямого секвенирования экзона 7а гена FGFR1. В других случаях выполняют секвенирование 7-го и 9-го экзонов гена FGFR2 – именно там возникает большинство дефектов, приводящих к развитию синдрома Пфайффера. Вспомогательную роль в диагностике заболевания могут играть методы медицинской визуализации, направленные на определение аномалий развития внутренних органов.
Лечение синдрома Пфайффера только симптоматическое и паллиативное – специалисты стараются устранить повышенное внутричерепное давление, обеспечить нормальное развитие нервной системы, снизить выраженность нарушений со стороны внутренних органов. Для этого широко применяют хирургические методики – разделение пальцев, пластику черепа и ряд других. Также назначают препараты, направленные на улучшение питания нервной ткани – ноотропные средства, витамины. При своевременно начатом лечении синдрома Пфайффера 1-го типа выживаемость больных значительно увеличивается. К сожалению, объем медицинской помощи зачастую недостаточен для полноценного лечения больных синдромом Пфайффера 2-го и 3-го типов.
Прогноз и профилактика синдрома Пфайффера
Прогноз синдрома Пфайффера определяется вариантом этого заболевания. Первый тип характеризуется относительно благоприятным прогнозом, который зависит от своевременности выявления патологии и правильности подобранного лечения. В благоприятных условиях больные сохраняют нормальный интеллект, доживают до преклонного возраста и способны иметь потомство – при этом риск передачи заболевания детям составляет минимум 50%. Однако синдром Пфайффера 2-го и 3-го типов обладает крайне неблагоприятным прогнозом – совокупность неврологических и других нарушений часто становится причиной летального исхода в раннем детстве. Даже при выполнении паллиативных мероприятий у больных наблюдается выраженная олигофрения и глубокая инвалидизация. Профилактика синдрома Пфайффера, учитывая часто спонтанный механизм развития этого заболевания, возможна только в рамках пренатальной диагностики ультразвуковыми или генетическими методами.
Источник
Синдром Пфайффера — редкое генетическое состояние, которое вызывает преждевременное слияние черепа, что приводит к ненормальному формированию лица и головы. Это также влияет на руки и ноги.
Причиной синдрома Пфайфера является мутация генов, ответственных за развитие пренатальной кости. Эта мутация ускоряет развитие костей, в результате чего череп сливается преждевременно. Существует три подтипа синдрома Пфайфера, причем типы 2 и 3 являются самыми тяжелыми.
Лечение начинается с рождения, как только будет сделан точный диагноз. Никакие обработки не могут остановить синдром Pfeiffer, но лечение может управлять специфическими симптомами этого состояния.
Быстрые факты о синдроме Пфайффера:
- По словам, синдром Пфайфера поражает около 1 из 120 000 новорожденных.
- Синдром Пфайффера является результатом наследственной мутации аутосомного доминантного гена или новой мутации гена.
- Существует три подтипа синдрома Пфайфера, классифицированных по степени тяжести.
- Хирургия играет центральную роль в лечении синдрома Пфайфера.
Что такое синдром Пфайфера?

Как правило, кости черепа ребенка собирались вместе только после того, как голова достигла своего полного размера. Но в случае синдрома Пфайфера пластины сближаются слишком рано, и череп не может расширяться во времени с растущим мозгом, что вызывает ненормальное формирование лица и головы.
Младенцы, рожденные синдромом Пфайфера, могут также иметь выпуклые глаза, высокие лбы, битые носы и затонувшие средние лица. Их пальцы и пальцы ног могут быть перепончатыми или короткими и широкими.
Причины и факторы риска
Автосомальные генетические нарушения требуют только одной копии атипичного гена, чтобы вызвать расстройство. Атипичный ген может быть унаследован от одного родителя или может быть результатом новой мутации гена у ребенка.
Большинство людей с синдромом Пфайфера развивают его из новой мутации, потому что ни у одного из родителей не существует мутации гена, которая может быть передана. Согласно Национальной Ассоциации Craniofacial, родитель с синдромом Пфайфера имеет 50-процентный шанс передать условие своему ребенку.
Подтипы синдрома Пфайфера
Существует 3 подтипа синдрома Пфайфера:
Тип 1
Тип 1 синдром Pfeiffer характеризуется преждевременным слиянием черепов, аномалий пальцев и пальцев стопы и затонувшими скулами. Неврологическое развитие ребенка и интеллектуальные способности обычно совпадают с другими детьми.
Люди с синдромом Pfeiffer типа 1 могут иметь нарастание жидкости в мозге и потерю слуха. Поскольку тип 1 представляет собой более мягкую форму синдрома Пфайфера, индивидуумы с этой формой заболевания имеют нормальную продолжительность жизни при условии успешного лечения.
Тип 2

Согласно отчету, у людей с синдромом Пфайфера типа 2 есть черепа черепа, похожие на черепа, в результате чрезмерного слияния черепов.
Также могут быть:
- аномальные глазные выступы, которые могут влиять на зрение
- слитые локтевые суставы
- плавленые коленные суставы
- пальцевые аномалии
- аномалии ног
- задержки развития
- неврологические осложнения
Тип 2 вызывает тяжелые неврологические дефициты, имеет плохой прогноз и часто приводит к ранней смерти.
Тип 3
Тип 3 синдром Pfeiffer вызывает те же виды инвалидности, что и тип 2, за исключением черепа cloverleaf. Перспективы людей с синдромом Пфайфера типа 3 также часто являются бедными и могут привести к ранней смерти.
симптомы
В дополнение к физическим нарушениям, включая слияние черепа, слитые локоть и коленные суставы, а также инвалидность пальцев и ног, синдром Пфайфера может также вызывать следующие симптомы:
- выпуклые или широко раскрытые глаза
- высокий лоб
- острый нос
- недоразвитость или чрезмерное развитие челюстей
- стоматологические проблемы
- потеря слуха
- проблемы развития головного мозга и другие неврологические дефициты в типах 2 и 3
- задержки развития в типах 2 и 3
Симптомы различны у индивидуумов.
диагностика
Диагноз синдрома Пфайфера производится с помощью исследований изображений и физического осмотра, чтобы подтвердить наличие преждевременных слияний кости в черепе, слитых локтевых и коленных суставах, а также нарушениях пальцев и ног.
Другие генетические условия, возможно, должны быть исключены, и врачи обычно проводят молекулярно-генетическое тестирование для подтверждения мутаций гена.
Каковы варианты лечения?

Дети с синдромом Пфайффера часто переносят несколько сложных операций на восстановление черепов и деформаций суставов.
Хирургия для выпуска преждевременно слитого черепа проводится в течение первого года жизни ребенка, чтобы способствовать нормальному росту мозга и черепа.
Хирурги также могут восстанавливать глазные гнезда ребенка одновременно, чтобы сохранить свое видение. Другие операции на лицевой поверхности, включая скулы и челюсти, а также операции на перепончатых руках и ногах ребенка, выполняются, когда ребенок старше.
Некоторым детям потребуется лечение, чтобы справиться с проблемами дыхания, что может включать:
- Хирургия, чтобы высвободить завалы середины лица.
- Хирургия для удаления миндалин или аденоидов (железы, расположенные на крыше рта, которые защищают от инфекции).
- Непрерывная положительная терапия давления в дыхательных путях (CPAP) с использованием специальной маски во время сна.
- Трахеостомия проводится в тяжелых случаях. Трахеостомия — это хирургическое отверстие через переднюю часть шеи и в трахею (трубу).
Некоторым детям может потребоваться стоматологическая работа по ремонту зубов и подложек. Другие могут нуждаться в речевой и языковой терапии.
Еда на вынос и перспективы
Люди с синдромом типа 1 Pfeiffer могут иметь нормальные сроки жизни, при условии, что они не страдают от осложнений заболевания и проходят успешное лечение.
Люди с типами 2 и 3 имеют тяжелые формы этого расстройства и имеют тенденцию к сокращению продолжительности жизни из-за проблем с дыханием и неврологических осложнений.
Эффективное лечение, как правило, вращается вокруг хирургии.
Источник
Синдром Пфайффера — чрезвычайно редкое генетическое заболевание, которое встречается в среднем у одного из 100 тыс. новорожденных. Одинаково часто поражает мальчиков и девочек. Основной симптом болезни — раннее сращивание костей черепа в процессе эмбриогенеза, из-за чего мозг не может в дальнейшем нормально развиваться.
Общие сведения
Заболевание было открыто известным немецким генетиком Рудольфом Пфайффером. В 1964 году он описал симптомы ранее неизвестной болезни, основными признаками которой были аномалии в структуре черепа и сращивание пальцев на ступнях или кистях рук (синдактилия). Синдром наблюдался у восьми пациентов из одной семьи, что говорило о генетической природе болезни. Дальнейшие исследования показали, что синдром Пфайффера передается по аутосомно-доминантному типу.
Ультразвуковое исследование позволяет обнаружить это заболевание у плода уже во втором триместре беременности. На УЗИ видны аномалии развития черепа, внутренних органов и конечностей.
Симптомы
В 1993 году американский генетик Майкл Коэн предложил классифицировать синдром Пфайффера на три типа в зависимости от тяжести симптомов. Сейчас его классификация является общепринятой и широко используется в медицинской литературе.
Симптомы синдрома Пфайффера 1 типа включают краниосиностоз — состояние, при котором один или несколько черепных швов преждевременно сливаются, образуя целостную кость. Как результат отмечаются изменения в форме черепа и аномальные черты лица — гипертелоризм, низко посаженные уши. Может иметь место ухудшение зрения, повышенное внутричерепное давление, расщепление неба. У 50% пациентов отмечается снижение слуха. Большинство людей с данным типом болезни обладают нормальным уровнем интеллекта и не имеют неврологических отклонений.
У детей с типом 1 синдрома Пфайффера внешние признаки, такие как широкие короткие пальцы на руках и ногах, наблюдаются почти всегда. Синдактилия — возможный, но не обязательный симптом для этого типа патологии.
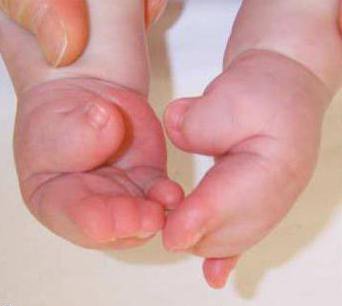
Данный тип синдрома наследуется аутосомно-доминантно.
Ниже представлено фото синдрома Пфайффера. Продолжительность жизни в России для людей, страдающих этим заболеванием, превышает 60 лет.
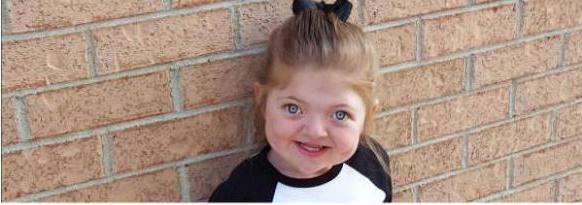
Для заболевания 2-го типа характерен череп в форме «трилистника», слияние позвонков, синдактилия и смещение глазных яблок вперед (проптоз). С первых дней жизни проявляются серьезные неврологические нарушения.
3-й тип заболевания характеризуется еще более ранним краниосиностозом. Череп аномально вытянутый и не образует «трилистник». Имеют место аномалии зубов, гипертелоризм, умственная отсталость, пороки развития внутренних органов.
Продолжительность жизни при 2-м и 3-м типе заболевания составляет от нескольких дней до нескольких месяцев — пороки внутренних органов и неврологические нарушения не совместимы с жизнью. Мутации, отвечающие за эти два типа синдрома Пфайффера, возникают спорадически и генетически не наследуются.
Причины
Синдром Пфайффера связан с мутациями рецептора 1 фактора роста фибробластов (FGFR1) на хромосоме 8 или с рецептором фактора роста фибробластов 2 (FGFR2) на хромосоме 10. Гены, которые затрагивают мутации, отвечают за нормальный рост фибробластов, что играет ключевую роль в развитии костей организма.
Предполагается, что одним из факторов риска 2-го и 3-го типа синдрома является возраст отца из-за увеличения числа мутаций в сперме с годами.
Лечение
Современная медицина пока не позволяет устранить мутации в человеческом геноме, даже если известно, в каких именно генах произошли перестройки. Поэтому для синдрома Пфайффера, как для большинства других генетических заболеваний, лечение является симптоматическим. Для пациентов со 2-м и 3-м типом заболевания прогноз неблагоприятный: многочисленные аномалии развития не могут быть скорректированы ни медикаментозно, ни хирургически.
Синдром Пфайффера 1 типа совместим с жизнью. Основной симптом заболевания — преждевременное сращивание костей черепа — может быть скорректирован хирургически. Операцию рекомендуется проводить детям до достижения трехмесячного возраста.
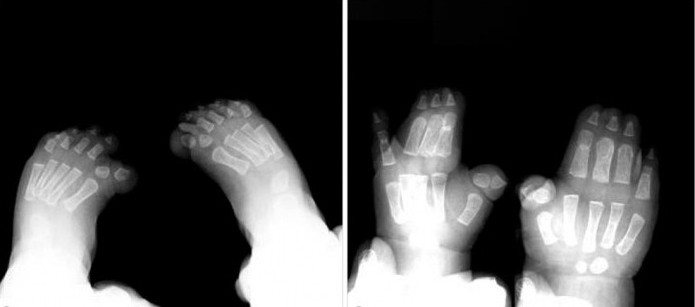
Кроме того, хирургическое вмешательство является эффективным в случае простой синдактилии. Лечение лучше всего начинать в возрасте 18-24 месяцев, в период активного роста пальцев на руках и ногах.
Источник