Синдром кляйнфельтера и синдром марфана

Синдром Клайнфельтера
Мужчины с так им синдромом обладают добавочной X хромосомой. Их набор половых хромосом — XXY. Исследования показывают, что лишнюю X хромосому они получают практически с равной вероятностью либо от матери, либо от отца, причем с увеличением возраста отца вероятность такою «сбоя» возрастает. Частота рождений детей с подобным хромосомным нарушением довольно высока, она составляет около 1/500. При этом до начала полового созревания обычно никаких отклонений от нормы при чисто внешнем осмотре ребенка обнаружить не удается, хотя при цитологических исследованиях у таких мальчиков в клетках обычно четко выявляется характерное для женского пола тельце Барра. В процессе возмужания у больных с этим синдромом складывается евнухоидный тип строения тела: узкие плечи и грудная клетка, широкий таз, слабо развитая мускулатура и волосяной покров на лобке и под мышками. Семенные канальцы часто атрофируются, а сперматозоиды не вырабатываются, что является причиной стерильности. У мужчин с синдромом Клайнфельтера регистрируется повышенный уровень характерного для женщин фолликулостимулирующего гормона, который выделяется с мочой. Молочные железы у них начинают увеличиваться (гинекомастия), что отмечал еще X. Клайнфельтер, впервые в 1942 г. описавший этот синдром. Если быть точным, то настоящими женскими молочными железами такую увеличенную мужскую грудь, впрочем, считать нельзя, поскольку она состоит из плотной соединительной ткани, которая не способна к лактации (к выделению молока).
Люди с синдромом Клайнфельтера обычно безынициативны и редко способны к творческой деятельности. Они легко поддаются внушению и эмоционально неустойчивы. Интеллект нередко при этом не страдает, хотя в некоторых случаях отмечается задержка умственного развития, достигающая порой дебильности. Около 15 % пациентов с синдромом Клайнфельтера страдают олигофренией. Некоторые замыкающиеся в себе дети, не реагирующие адекватно на внешний мир (аутисты), нередко при обследовании оказываются обладателями хромосомного набора XXY.
Почти всегда умственная отсталость выявляется у больных с хромосомным набором XXXY или даже с XXXXY. Внешне таких людей можно четко идентифицировать как мужчин, однако они стерильны и обладают внешностью евнухов. Несколько сгладить проявление синдрома Клайнфельтера можно с помощью инъекций аналога мужского полового гормона метилтестостерона, которые врачи рекомендуют начинать делать в возрасте 10–11 лет. Поэтому очень важно вовремя идентифицировать таких больных, что можно сделать в результате анализа их клеток.
Следующая глава >
Похожие главы из других книг:
Синдром Шершевского — Тернера
Отсутствие одной из хромосом диплоидного набора в генетике называется моносомией (греч. monos — один). В этом случае в клетках присутствует лишь одна из двух гомологичных хромосом. В подавляющем большинстве случаев зародыши с такой аномалией
Адреногенитальный синдром
Как известно, надпочечники позвоночных животных и человека вырабатывают несколько очень важных гормонов, среди которых различают адреналин, мужские половые гормоны андрогены (греч. andros — мужчина) и так называемые кортикостероиды. Последние
Синдром Дауна
Во второй половине XIX века на протяжении более десяти лет Дж. Лангдон-Даун — английский врач психиатрической лечебницы в графстве Суррей — наблюдал за умственно отсталой девочкой. У нее было плоское лицо с широкой переносицей и приплюснутым носом, пухлые
Синдром кошачьего крика
В 1963 г. французский исследователь Лежен описал у новорожденных детей врожденную аномалию, которую он назвал синдромом «кошачьего крика». Дело в том, что страдающие им дети имели мяукающий тембр голоса. Такая необычная особенность определялась
Синдром Марфана
Секреты Андерсена, Паганини и Чуковского
Дефект некоторых генов, влияющих на образование и развитие соединительной ткани у человека, нередко приводит к непропорциональному гигантизму. При наиболее ярком проявлении этой доминантной особенности на свет
Синдром «Фольксвагена»
На ранний и начало позднего протерозоя (2,5–1,5 млрд лет назад) пришелся своеобразный застой. Ничего откровенно нового в течение долгих полутора миллиардов лет не появилось. Даже изотопная летопись углерода имеет вид скучной прямой линии без
Синдром Суайра
Человеческий эмбрион с кариотипом XY начинает становиться мужчиной только после шестой недели своего развития при условии достаточного количества мужских гормонов. Если вдруг по каким-либо причинам в Y-хромосоме зачатого «мальчукового» эмбриона
Синдром де ля Шапеля
Один из 25 000 мужчин на самом деле женщина. Их кариотип представлен женским набором хромосом – ХХ.Внешне такие мужчины мало отличаются от обычных. Они способны к половой жизни, хотя у них часто понижено либидо и они бесплодны. У мужчин, страдающих
Синдром Кляйнфельтера
Может случиться так, что во время оплодотворения в яйцеклетку проникнет «мальчуковый» сперматозоид, несущий в себе одну (или даже больше) дополнительную Х-хромосому. Тогда гаметный набор будет выглядеть как XXY, а количество хромосом в наборе будет
Синдром Марфана
Синдром Марфана, особая форма диспропорционального гигантизма, — результат системного дефекта соединительной ткани; наследуется доминантно, т. е. по вертикальной линии, но с очень варьирующими проявлениями. При полном проявлении наблюдаются: высокий
12.2. Болезнь Клайнфельтера как причина пассивной антисоциальности
Совершенно иного типа преступность связана с синдромом Клайнфельтера, характеризующимся наличием 47.хромосом, в том числе набором половых хромосом XXY, недоразвитием семенников, евнухоидным телосложением,
13.4. Синдром убийства королей и президентов
Нельзя оставить без внимания также и опыт мировой истории, ясно демонстрирующий, какую роль могут играть паранойяльные личности со своего рода геростратовым комплексом или стремлением отомстить обществу за свои личные
7.1. «Синдром общих блоков»
Концепция универсальных функциональных блоков вводит нас в новую область медицины, относящуюся к молекулярным заболеваниям. В настоящее время уже обнаружены заболевания, которые должны быть истолкованы с позиций этой концепции. Такие
Пикквикский синдром
Появление быстрого сна сразу после засыпания свойственно не одной нарколепсии, но и некоторым другим заболеваниям. Да и здоровый человек, проснувшись среди быстрого сна, может заснуть и опять очутиться в быстром сне. Чтобы попасть в быстрый сон, не
Источник
Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.

Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.

Симптомы синдрома Марфана
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
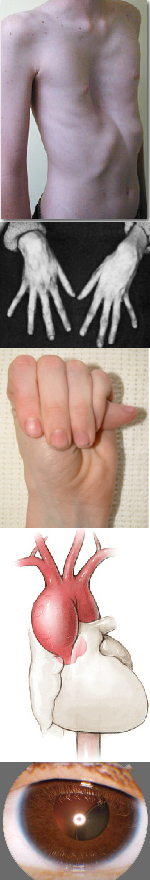 Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз, сколиоз;
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— плоскостопие;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— высокое нёбо;
— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.

Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:

Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн. Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Источники и ссылки:
1. Причины высокого роста — статья, в которой рассмотрены основные причины высокого роста человека.
2. Синдром Марфана в википедии — о синдроме Марфана в свободной энциклопедии.
3. Русскоговорящее сообщество людей с синдромом Марфана — форум о синдроме Марфана, общение, обсуждение болезни.
4. Международный фонд синдрома Марфана — фонд помощи больным с синдромом Марфана.
5. Группа вконтакте для больных синдромом Марфана.
Источник
Никколо Паганини
О том, что великий скрипач был болен синдромом Марфана, стало известно лишь впоследствии, в 1978 году, после выхода работы* авторства Мирона Р. Шёнфилда «Никколо Паганини: музыкальный волшебник или мутант с синдромом Марфана?» (Nicolo Paganini: Musical Magician and Marfan Mutant?). Интересно, что автор, чьим именем названа патология, на самом деле описал немного иное расстройство, похожее на него, и сделал это отнюдь не первым. Его на 20 лет опередил врач-офтальмолог.
E. Williams многие годы занимался глазными заболеваниями, и однажды к нему на прием в июле 1875 года пришли сестра и брат, Мэри и Маркус Дж. 28 летняя девушка хотела проконсультироваться с врачом насчет чего-то непонятного, произошедшего с ее глазами. Врач обнаружил, что хрусталик ее правого глаза сместился из зоны зрачка на ¼ вверх, а хрусталик левого, тоже смещенного вверх, оставлял зону зрачка на 2/3 пустой.
Ее брат, который был младше на 2 года, никогда не страдал какими-то расстройствами, к докторам не обращался и всегда имел превосходное зрение. К врачу он пришел вместе с сестрой просто за компанию, однако, от зоркого взгляда Вильямса не укрылась странность расположения его хрусталиков, не столь выраженная, как у сестры, но все-таки имеющаяся.
В итоге офтальмолог обнаружил, что у обоих произошла спонтанная эктопия (смещение положения) хрусталика. При этом он обратил внимание на то, что оба были очень высокого роста, худощавые и с детства имели слишком подвижные суставы. Врач описал эти случаи в своей работе** «Rare Cases, with Practical Remarks» и не смог их как-либо интерпретировать, кроме как симптоматически.
Через 20 лет после этого в 1896 году интересный случай пятилетней девочки Габриэллы П. отметил Антуан Бернард-Жан Марфан. Он в те годы работал детским врачом в Société Médicale des Hôpitaux de Paris и в возрасте 38 лет уже имел степень доктора медицинских наук, общественное признание и доверие пациентов.
Антуан Марфан
Но прежде чем рассказывать о случае, давайте поговорим о самом Антуане Марфане. Он родился в 1858 году в семье сельского врача, который весьма скромно зарабатывал, трудно жил и поэтому отговаривал своего сына от медицины, советуя ему поступать в Эколь Политекник. Однако, совладать с упорным юношей не получилось, и Марфан поступил в медицинскую школу в Тулузе. После проведенных там двух лет он отправился в 1879 году в Париж, а уже в 1880 получил диплом и начал практику.
В 1892 году он в зимние месяцы работал в клинике Hôpital des Enfants Malades и именно здесь увлекся педиатрией, с которой впоследствии свяжется его жизнь. Успел он поработать в разных «весовых категориях»: помощником профессора педиатрии на факультете медицины Парижского университета, главой службы по дифтерии в Больнице для больных детей (Hospital for Sick Children), первым профессором детской гигиены в детской поликлинике при Парижском университете.
Антуан Марфан серьезно занимался туберкулезом. Тема его докторской диссертации звучала как «Troubles and gastric lesions in pulmonary tuberculosis» или «Патологии и поражения желудка при туберкулезе легких». Кстати, этот документ дал основу концепции, известной сейчас как «закон Марфана» и говорящей о том, что легочный туберкулез в полной форме развивается довольно редко, если пациент переболел его локализованными формами, так как формируется сильный иммунитет. Исследования, подобные этому, как раз привели к разработке вакцины БЦЖ.
Помимо этого, детский врач был одним из первых, кто осознал важность наблюдений за реакциями кожи, и когда фон Пирке разработал свою методику кожных проб на туберкулез, он тут же применил ее в своих клинических исследованиях, которые впоследствии стали классикой фтизиатрии. Об этом мы подробнее расскажем в главе, специально посвященной этой теме.
Марфан стал одним из самых выдающихся педиатров Франции, в 1897 году он вошел в коллектив авторов Трактата детских болезней, который удостоился премии Французской академии наук. Он изучал вред от кормления младенцев козьим молоком, провел обширнейшие исследования на тему рахита. И, конечно же, он имел дело с Жаном-Мартеном Шарко, причем, довольно почетное: делил должность редактора в журнале System of Medicine, а также сам основал журнал Le Nourrisson (Младенец). Неудивительно, что Марфана вообще считают одним из создателей педиатрии как таковой.
Жан-Мартен Шарко
Марфан прожил 84 года и оставил нам несколько медицинских названий. Например, синдром Денни-Марфана (вариан врожденного сифилиса у младенцев, проявляющийся в спастическом параличе верхних или нижних конечностей и умстевенной отсталости), описанный им независимо от американца Чарльза Клейтона Денни, или закон Марфана, который относится к лечению туберкулеза и говорит о том, что излечение очагового (вторичного) туберкулеза легких защищает от развития общего туберкулеза легких.
Педиатр говорил: «в медицине всегда необходимо начинать с наблюдения за больными и всегда возвращаться к этому, поскольку это является первостепенным средством проверки. Наблюдайте методично и энергично, не пренебрегая какой-либо исследовательской процедурой, используя все, что может быть охвачено с помощью физического осмотра, химических, бактериологических исследований и эксперимента, так как необходимо сравнить факты, наблюдаемые в течение жизни, и выявленные открытия».
Наверное, именно это качество помогло ему обратить внимание на маленькую и ни на кого не похожую по симптомам Габриэллу.
В статье*** «A case of congenital deformation of the four limbs, more pronounced at the extremities, characterized by elongation of the bones with some degree of thinning» он дал весьма подробное описание ребенка, который имел непропорционально длинные конечности и астеничное телосложение. Но более всего врача привлекали ее пальцы на руках и ногах, которые были похожи на лапки паука – столь же длинные и тонкие. Он даже назвал этот феномен «pattes d’araignée», а саму патологию – долихостеномелия (с греческого «stenos» — стрела, копье, а «melos» — конечность).
Кисть больного синдромом Марфана
К сожалению, девочка прожила не очень долго и умерла в подростковом возрасте от туберкулеза, против которого на тот момент еще не было какого-либо специфического лечения. Но Марфан наблюдал ее до самой смерти и отмечал прогрессирование странных и искренне поразивших его аномалий.
Однако, как чуть позже выяснили Анри Мери и Леон Бабонне, осмотревшие ребенка в 1902 году с помощью новейшего метода визуализации – рентгена, который только начал появляться в клиниках, у нее наблюдалась арахнодактилия, изменения грудной клетки и позвоночника не из-за ахондроплазии, как предположил Марфан, а из-за гиперхондроплазии – чрезмерного образования хрящевой ткани. В том же 1902 году описание подобных пациентов появилось еще от одного врача, Ашара. Оно углубилось более внимательными фактам о патологии сердечно-сосудистой системы в виде перерастяжения и расслоения аорты, о вывихе хрусталика и наследственном характере всех этих признаков.
Вверху: большой палец в норме, внизу — при синдроме Марфана
Но все эти исследователи пока не подозревали, что первый случай – описание Гарбиэллы – это не тот самый «синдром Марфана», которым начали называть все похожие варианты. Скорее, она страдала врожденной контрактурной арахнодактилией, которая относится к заболеваниям соединительной ткани, но не имеет тот тип наследования, который характерен для синдрома. Естественно, это все выяснилось гораздо позже, лишь во второй половине 20 века.
Ну и напоследок нам стоит развеять еще один миф. Очень часто «самым известным носителем синдрома Марфана» называют одного из самых распиаренных правителей древности – фараона Тутанхамона. Этот фараон правил в XIV веке до нашей эры, занял трон в возрасте десяти лет и правил почти столько же. Исходя из его изображений «исследователи» достаточно часто «ставили» Тутанхамону синдром Марфана (и еще десяток других заболеваний). Однако недавнее исследование мумии фараона методом компьютерной томографии и секвенирование генома Тутанхамона показали, что юноша имел целый букет заболеваний — волчья пасть (врожденное незаращение твердого неба и верхней челюсти), косолапость, болезнь Кёлера (деформации ступней, вызванные нарушением кровоснабжения отдельных костей стопы), но вот телосложение у него было абсолютно нормальное. Так что синдромом Марфана тут и не пахло. А вот от чего умер этот юноша вы узнаете, прочитав об этом фараоне в нашей новой книге, которая уже готовится к печати))).
Тутанхамон на колеснице
Следить за обновлениями нашего блога можно и через его страничку в фейсбуке и паблик
вконтакте
*Myron R. Schoenfeld, MD. Nicolo Paganini. Musical Magician and Marfan Mutant? JAMA. 1978;239(1):40-42. doi:10.1001/jama.1978.03280280040022
**Williams E. Rare Cases, with Practical Remarks // Transactions of the American Ophthalmological Societ. — 1875. — Vol. 2. — PP. 291—301. — PMC 1361735
***Marfan, Antoine (1896). Un cas de déformation congénitale des quartre membres, plus prononcée aux extrémitiés, caractérisée par l’allongement des os avec un certain degré d’amincissement [A case of congenital deformation of the four limbs, more pronounced at the extremities, characterized by elongation of the bones with some degree of thinning]. Bulletins et memoires de la Société medicale des hôspitaux de Paris. 13 (3rd series): 220–226.
Источник