Реферат на тему синдром орбели

Дети с синдромом Орбели имеют интеллектуальную инвалидность и врожденные пороки развития. Их тяжесть и доступные методы лечения, как диагностируется заболевание при беременности и после рождения, прогноз качества и продолжительности жизни — информация для родителей.
В этой статье:
- Что такое синдром Орбели?
- Фото больных
- Симптомы
- Клинический случай
Что такое синдром Орбели?
Синдром Орбели — это врожденное генетическое заболевание, возникающее при отсутствии (удалении) генетического материала на длинном плече 13 хромосомы. Тяжесть состояния и клиническая картина зависят от количества удаленных генов и их расположения.
Спектр нарушений, характерных для детей с удалением сегмента 13 хромосомы, включает задержку развития, умственную отсталость, поведенческие проблемы и особенности внешности. Болезнь Орбели также называют делецией или моносомией длинного плеча 13 хромосомы.
Тест кариотипа (анализ крови) обоих родителей покажет, была ли делеция унаследована. У большинства родителей детей с синдромом Орбели хромосомная аномалия отсутствует, но у одного из тестируемых может обнаружиться сбалансированная транслокация (разрыв участка хромосомы или присоединение к другой хромосоме с потерей генетического материала или без такого). Сбалансированная транслокация не вызывает симптомов, но увеличивает риск рождения младенца с генетическими аномалиями.
Лечение зависит от признаков синдрома у ребенка и подбирается индивидуально.
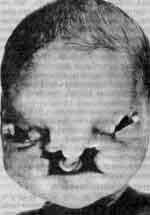
Фото больных
При синдроме Орбели дети имеют специфическую внешность и сочетанные дефекты развития. Фото дают представление о заболевании.
Симптомы
Признаки очевидны при рождении, клиническая картина дополняется по мере взросления. Из первых и самых распространенных симптомов выделятся низкий вес новорожденного, пороки развития черепно-лицевого скелета, глаз, конечностей, половых органов и другие физические аномалии.
У всех детей с диагнозом синдром Орбели наблюдается задержка психомоторного развития: с трудом дается приобретение навыков, требующих умственной и физической активности. Обязательно наличие умственной неполноценности различной степени выраженности.
Задержка роста от умеренной до тяжелой может наблюдаться в любом возрасте или сопровождать весь процесс взросления, что приведет к низкорослости.
Для синдрома Орбели характерны многие аномалии строения лица и черепа (необязательно сопровождают заболевание и сами по себе не являются его признаком):
- микроцефалия (размер мозга ниже нормы, маленькая голова);
- тригоноцефалия (ранее закрытие метопического шва, соединяющего две половины лобной кости, треугольная голова);
- широкий и плоский носовой мост;
- большие, искривленные или низко расположенные уши;
- микрогнатия (недоразвитая, маленькая нижняя челюсть) с аномально выраженной верхней челюстью;
- дистальный прикус (выпирают передние зубы);
- волчья пасть (расщелина неба);
- заячья губа (расщелина верхней губы);
- крыловидная шея (кожные складки с обоих сторон шеи протяженностью до предплечья).
Возможные нарушения строения головного мозга:
- эпилепсия;
- гидроцефалия (скопление спинномозговой жидкости в ликворной системе);
- голопрозэнцефалия (мозг не разделен на полушария);
- менингоцеле (мозговая грыжа);
- отсутствие частей мозга, например, агенезия мозолистого тела или обонятельного отдела;
- гипоплазия (недоразвитие) или агенезия (отсутствие) мозжечка;
- синдром Денди Уокера.
Дефекты развития нервной трубки у плода (при синдроме Орбели встречаются крайне редко, обычно в сочетании с другими генетическими заболеваниями):
- анэнцефалия;
- энцефалоцеле;
- расщепление позвоночника.
Аномалии развития глаза (возможные варианты):
- микрофтальмия (маленький размер и деформация одного или обоих глазных яблок);
- широко посаженные глаза;
- опущение верхнего века;
- эпикантальные (монгольские) складки, закрывающие уголки глаз.
- катаракта;
- помутнение роговицы;
- отсутствие радужной оболочки.
Нарушения строения глаза при синдроме Орбели влияют на зрение, редко вызывают слепоту.
Пороки развития конечностей:
- гипоплазия (недоразвитие) или отсутствие больших пальцев;
- мизинец скошен внутрь;
- слияние пальцев;
- короткие пальцы;
- короткие ноги;
- скошенные стопы (косолапость).
Пороки развития скелета:
- аномалии развития ребер и/или позвоночных костей;
- сколиоз (боковое искривление позвоночника).
Генитальные аномалии часто встречаются у мальчиков и характеризуют синдром Орбели в медицинской литературе:
- недоразвитие пениса;
- смещение отверстия мочеиспускательного канала вниз;
- неопущение яичка в мошонку;
- маленькая мошонка;
- двойная мошонка;
- микропенис.
У девочек возможны неправильное развитие, гипоплазия, отсутствие женских половых органов.
У обоих полов:
- промежностный свищ;
- стеноз или отсутствие анального отверстия.
Аномалии внутренних органов у детей с синдромом Орбели:
- врожденные предсердные и желудочковые дефекты сердца;
- открытое овальное окно в сердце (часто не проявляется, но может спровоцировать худобу, мягкую задержку роста, пониженную сопротивляемость к респираторным инфекциям, одышку, легкую утомляемость при физических нагрузках, аритмию);
- дефект межжелудочковой перегородки (малый может закрыться самостоятельно, умеренный спровоцирует застойную сердечную недостаточность);
- гипоплазия, эктопия, поликистоз или отсутствие почки;
- дефекты легких.
Тяжелые дефекты строения внутренних органов могут привести к смерти новорожденного.
Основные симптомы всех видов частичной делеции длинного плеча 13 хромосомы и их частотность в процентах.
| Признаки | Количество затронутых детей (%) |
|---|---|
| Низкий вес новорожденного (меньше 2,5 кг) | 45 |
| Задержка роста | 30 |
| Психомоторная отсталость | 100 |
| Микроцефалия | 30 |
| Брахицефалия | 75 |
| Тригоноцефалия | 20 |
| Асимметрия лица | 30 |
| Гипертелоризм | 45 |
| Аномалии глаз | 75 |
| Колобома (отсутствие фрагмента оболочки глаза) | 15 |
| Аномальное строение носового моста | 80 |
| Большие или неправильно сформированные уши | 85 |
| Патологии строения челюсти | 55 |
| Аномалии строения и работы сердца | 30 |
| Врожденный порок сердца | 20 |
| Аномалии почек | 40 |
| Генитальные аномалии | 45 |
| Анальная атрезия | 7 |
Клинический случай
Ребенок женского пола, родился на 38 неделе беременности, гестационный период осложнен внутриутробной задержкой роста.
Показатели при рождении:
вес — 2400 г;
рост — 42 см;
окружность головы — 31 см.
Данные осмотра:
- отсутствие сосательного рефлекса;
- микроцефалия;
- гипертелоризм;
- катаракта;
- выступающий носовой мостик;
- короткий губной желобок;
- высокое небо;
- короткие пястные кости;
- на левой руке 4 пальца, на правой 5 с гипоплазией большого пальца;
Другие замечания:
- мышечный гипотонус;
- незаращение дужки 3 позвонка;
- гипоплазия мозолистого тела;
- гипоплазия обоих почек;
- дефект межпредсердной и межжелудочковой перегородок, открытие артериального протока;
- маленькие уши неправильной формы;
- неполное прикрепление половых губ;
- отсутствие матки и яичников.
Описан тяжелый случай синдрома Орбели.
Причины
Хромосомы находятся в ядре каждой клетки, несут генетические характеристики. Каждая хромосома имеет короткое плечо и длинное плечо. Далее они подразделяются на полосы (участки, сегменты), которые нумеруются. Например, 22 участок 13 хромосомы.
В случае частичной делеции, то есть удаления сегмента длинного плеча 13 хромосомы обязательно наступают последствия для развития и здоровья ребенка: от малозначительных до глобальных. Понятие синдрома Орбели охватывает делецию проксимальных (центральных) полос длинного плеча 13 хромосомы: с 22 по 31 сегмент. При удалении полос хромосомы в этих пределах наблюдается задержка роста, физические отклонения и умственная неполноценность различной степени тяжести.
Дети с делецией проксимальных участков имеют другие симптомы (не входят в спектр нарушений при синдроме Орбели). Делеция 14 участка создает повышенный риск ретинобластомы (злокачественное новообразование сетчатки). Если затронут 32 сегмент 13 хромосомы, ожидается тяжелая умственная отсталость и другие пороки по аналогии с синдромом Орбели. Делеции 33-34 участка характеризуются изолированными интеллектуальными недостатками и в редких случаях черепно-лицевыми аномалиями, обычно не связаны с другими синдромальными пороками развития. Олигофрения (низкий интеллект) может сопровождаться поведенческими отклонениями или расстройствами аутического спектра.
В большинстве описанных случаев синдрома Орбели исследователи полагают, что делеция спровоцирована нарушениями на начальном этапе эмбрионального развития. В таких случаях родители больного ребенка обычно имеют нормальный кариотип, риск родить второго ребенка с подобной аномалией у них низкий.
Иногда частичная делеция 13 хромосомы спровоцирована хромосомной перестройкой у родителей. Возможна транслокация (смещение и перегруппировка), инверсия (разрыв и присоединение участка в обратном порядке) участков хромосомы. Такие перестановки безвредны для носителя, но могут отразиться на хромосомной организации у потомства.
Диагностика
Диагноз синдрома Орбели можно определить у ребенка пренатально, используя такие тесты:
- УЗИ (нейросонография), пренатальные МРТ и КТ показывают физические аномалии, характерные для генетического расстройства;
- амниоцентез (изучение образца жидкости, взятого из околоплодных вод);
- выбор хорионических ворсинок (исследование образца ткани, взятого из плаценты).
Подтвердить диагноз можно только после рождения с помощью клинического осмотра и анализа крови на хромосомную патологию.
Родители могут сдать анализ на кариотип и пройти генетическое консультирование.
Лечение
Планированием курса терапии и оказанием помощи ребенку занимаются врачи нескольких специальностей: педиатр, невролог, офтальмолог, кардиолог, радиолог, ортопед, хирург, онколог.
В некоторых случаях требуется проведение экстренной хирургической операции для коррекции физических пороков развития. Например, операция может потребоваться, чтобы устранить черепно-лицевые, глазные, генитальные пороки развития и дефекты сердца, связанные с синдромом Орбели или сопутствующими нарушениями, чтобы ребенок мог продолжать жизнедеятельность.
Проблемы со зрением и строением глаз решаются подбором коррекционных линз или очков, хирургической операцией.
При наличии тригоноцефалии или других форм краниостеноза рекомендовано проведение операции по коррекции деформаций свода черепа, причем желательно провести ее в раннем возрасте, пока доступна эндоскопическая хирургия (до 6-10 месяцев). Подробнее об этой аномалии и перспективах читайте в статье о краниостенозе и его формах.
Хирургически исправляется гидроцефалия: показана эндоскопия или шунтирование. Межжелудочковая перегородка корректируется хирургическим способом, но до операции проводят поддерживающее лечение сердечной недостаточности.
Лечение включает регулярный мониторинг неоперабельных нарушений. При ослабленном иммунитете и/или хроническом инфекционном процессе необходимо быстро начинать лечение даже незначительных (как кажется) респираторных заболеваний. Врачи рекомендуют вводить антибиотики перед каждой хирургической операцией, включая удаление зубов (для решения этого вопроса нужна консультация с несколькими специалистами, оценка рисков).
Дальнейшее ведение детей с синдромом Орбели предусматривает симптоматическую и поддерживающую терапию, чтобы помочь им достичь максимального потенциала в физическом и психическом развитии, социализироваться. Такой подход включает коррекционное образование, физиотерапию и другие методики развития.
Прогноз
Рост и взросление при синдроме Орбели осложнены множественными физическими пороками и олигофренией. В младенчестве возможны частые обмороки и судороги. Тщательное наблюдение за пациентом осуществляется, как минимум, до 8 лет.
Дети с тяжелыми пороками развития и развернутой клинической картиной не доживают до 1 года.
Выжившие люди имеют сокращенную продолжительность жизни. Максимальный возраст, зафиксированный у пациента с синдромом Орбели, составил 67 лет.
Большинству взрослых пациентов необходимы вспомогательные услуги для поддержания нормальной жизнедеятельности, включая работу сиделки и домработницы.
Источник
Частичная моносомия хромосомы –выпадение участка хромосомы. Как правило, возникают в результате структурных перестроек хромосом, имеющихся в половых клетках родителей, которые вследствие нарушения процессов рекомбинации в мейозе приводят к утрате или избытку фрагментов хромосом, вовлеченных в перестройку.
Частичные моносомии известны практически по всем хромосомам, но лишь некоторые из них формируют четко диагностируемые клинические синдромы. Фенотипические проявления этих синдромов более полиморфны, чем синдромов целых моносомий. Отчасти это связано с тем, что размеры фрагментов хромосом и, следовательно, их генный состав, могут варьировать в каждом отдельном случае, а также тем, что при наличии хромосомной транслокации у одного из родителей частичная моносомия по одной хромосоме у ребенка может сочетаться с частичной трисомией по другой.
Наиболее известные симптомы частичных моносомий – это Вольфа-Хиршхорна, «кошачьего крика», Орбели.
Синдром Вольфа-Хиршхорна
(4р-) обусловлен делецией короткого плеча четвёртой хромосомы. Частота заболевания составляет около одного случай на 100 тысяч. Болезнь характеризуется задержкой умственного и психомоторного развития. Также могут проявляться в большинстве случаев тяжелейшие пороки сердца, почек. У новорождённых небольшой вес при нормальной продолжительности беременности (до 2 кг). Среди внешних признаков могут отмечаться: микроцефалия, клювовидный нос, эпикант (складка у внутреннего угла глаза, в большей или меньшей степени прикрывающая слёзный бугорок), антимонголоидный разрез глаз (опущение наружных углов глазных щелей), аномальные ушные раковины, расщелина верхней губы и нёба, маленький рот, деформация стоп и др. Средняя продолжительность жизни примерно до 30 лет (в России зафиксирована максимальная продолжительность жизни 25 лет), при тяжёлых пороках сердца, почек продолжительность жизни может составлять не более одного года. Применяется симптоматическое лечение.
Синдром кошачьего крика
(5р-) обусловлен делецией короткого плеча пятой хромосомы. При этом синдроме наблюдается:общее отставание в развитии, низкая масса при рождении и мышечная гипотония, лунообразное лицо с широко расставленными глазами, характерный плач ребёнка, напоминающий кошачье мяуканье, причиной которого является изменение гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки) или недоразвитие гортани. Признак исчезает к концу первого года жизни. Кроме того, встречаются врожденные пороки сердца, костно-мышечной системы и внутренних органов, микроцефалия, птоз, низкое расположение и деформация ушных раковин, кожные складки впереди уха, гипертелоризм (увеличенное расстояние между какими-либо парными органами или анатомическим образованиями (например, между внутренними краями глазниц, грудными сосками), эпикантус (поперечная кожная складка около внутреннего угла глаза, обычно двусторонняя; наиболее чётко выражена при болезни Дауна, антимонголоидный разрез глаз. Частота синдрома примерно 1:45000. Соотношение полов М1 : Ж1,3. Клиническая картина синдрома и продолжительность жизни людей с этим синдромом довольно сильно варьирует по сочетанию врождённых пороков развития органов. Лечение симптоматическое. Показаны средства, стимулирующие психомоторное развитие, лечебный массаж и гимнастика.
Синдром Орбели
(13q-) обусловлен делецией длинного плеча тринадцатой хромосомы. Популяционная частота синдрома не установлена. Дети с синдромом Орбели рождаются с низким (2200 г) весом. Клинически синдром проявляется аномалиями развития всех систем организма. Характерны микроцефалия, отсутствие носовой вырезки (лоб непосредственно переходит в нос), эпикант, антимонголоидный разрез глаз, широкая спинка носа, высокое нёбо, низко расположенные деформированные ушные раковины. Отмечаются поражения глаз, опорно-двигательного аппарата (короткая шея, гипо- или аплазия первого пальца кисти и пяточной кости, синдактилии кистей и стоп), атрезии прямой кишки и заднепроходного отверстия. Часты пороки развития сердца, почек, головного мозга. Для всех детей с синдромом Орбели характерна глубокая олигофрения, возможны потери сознания и судороги. Большинство больных с синдромом 13q- погибают на 1-м году жизни.
Синдром Прадера-Вилли— редкая генетическая аномалия. При синдроме Прадера-Вилли отсутствуют примерно 7 генов из 15 хромосомы. При синдроме Прадера-Вилли страдает отцовская хромосома, в случае же повреждения материнской хромосомы возникает синдром Ангельмана. Для синдрома Прадера-Вилли характерны: до рождения: низкая подвижность плода, часто — неправильное положение плода, ожирение, склонность к перееданию, пониженный мышечный тонус, пониженная координация движений, маленькие кисти и стопы, низкий рост, повышенная сонливость, косоглазие, сколиоз, пониженная плотность костей, сниженная функция половых желёз, в результате, как правило, бесплодие, речевая задержка, задержка психического развития, отставание в освоении навыков общей и мелкой моторики, более позднее половое созревание. Внешние признаки: у взрослых выражена переносица, лоб высокий и узкий, глаза, как правило, миндалевидные, губы узкие. Частота встречаемости — 1 : 12000-15000 живорождённых младенцев. Синдром Прадера-Вилли является врожденной генетической аномалией и, следовательно, не может быть излечен.
Источник

Причина.
Делеция длинного плеча аутосомы 13.
Клиника.
Лоб переходит в нос, не образуя носовой
вырезки. Большое расстояние между
глазами. Широкая спинка носа, высокое
нёбо, низко расположенные диспластичные
ушные раковины, пороки развития глаз
(косоглазие, катаракта). Пороки
опорно-двигательного аппарата –
неспецифические аномалии (косолапость,
вывих тазобедренных суставов). Задержка
роста и психомоторного развития;
характерна
глубокая олигофрения.
Больные с развернутой клинической
картиной синдрома погибают на первом
году жизни.
Патогенез.
Аномальное развитие практически всех
органов и систем; микроцефалия; врожденные
пороки сердца и аномалии прямой кишки.
Диагностика.
Цитогенетическое, клиническое
обследование.
Лечение.
Отсутствует.
Частота.
Описано
более 100 пациентов с частичной моносомией
хромосомы 13 (синдром Орбели). Мальчики
и девочки — с одинаковой частотой.
Синдром “кошачьего крика”
Причина.
Делеция короткого плеча хромосомы 5-й
пары. Кариотип 46, 5р-.
Клиника.
Патологическое строение голосовых
связок – сужение, мягкость хрящей,
отечность и необычная складчатость
слизистой, уменьшение надгортанника,
поэтому дети издают крик, напоминающий
мяуканье кошки. Недоразвитие речи.
Микроцефалия. Лунообразное лицо,
монголоидный разрез глаз, косоглазие,
катаракта, атрофия зрительного нерва,
плоская спинка носа, высокое нёбо,
деформированные ушные раковины.
Косолапость. Задержка
умственного и физического развития.
Продолжительность жизни значительно
снижена, только около 14% больных переживают
возраст 10 лет.
Патогенез.
Порок сердца.
Диагностика.
Клиническое обследование с выявлением
наиболее постоянного признака синдрома
– “кошачий крик”, дерматоглифика и
цитогенетическое выявление патологии
кариотипа.
Лечение.
Отсутствует.
Частота.
1:50000.

Фронтазальная дисплазия (синдром срединной расщелины лица)
Причина.
большинство случаев спорадические.
Клиника.
глазной гипертелоризм, широкое основание
носа, умственная отсталость – 20% случаев
средней тяжести. Отличается клиновидный
рост волос на лбу («мыс вдовы»). Типичны
гипертелоризм и дефекты средних структур
черепа, варьирующие от скрытой расщелины
костей черепа до мозговой грыжи.
Патогенез.
3 формы
дисплозии (в зависимости от выраженности
расщелины костей черепа):
гипертелоризм,
широкое основание и скрытая расщелина
носа, иногда с раздвоением кончика
носа;гипертелоризм,
широкое основание носа, открытая
расщелина носа и губы. Возможна расщелина
нёба;тотальная
расщелина носа, отсутствие крыльев
носа, деформация глазниц.
В
ряде случаев встречаются брахицефалия,
микрофтальмия, эпикант, коломбы век,
врождённая катаракта, преаурикулярные
ушные раковины, иногда бывает проводящая
глухота, клинодактимия, крипторхизм,
камптодактимия, липомы дермоиды.
Отличаются сочетания фроитоназальной
дисплазии с гидроцефалией, амнезией
мозолистого тела. Встречается аномалия
пищевода.
Диагностика.
Клиническое обследование.
Лечение.
Хирургическое вмешательство.
Частота.
1: 1000000.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник