Прогноз при синдроме арнольда киари


Синдром Арнольда-Киари — врожденная патология краниовертебральной зоны, возникающая в период эмбриогенеза и характеризующаяся деформацией черепа. Опущение структур головного мозга в большое затылочное отверстие приводит к их сдавлению и ущемлению. Дисфункция мозжечка сопровождается нистагмом, шаткой походкой, дискоординацией движений, а поражение продолговатого мозга — изменениями в работе жизненно важных органов и систем.
Впервые синдром был описан в конце 18 века двумя учеными: доктором из Германии Арнольдом Джулиусом и врачом из Австрии Гансом Киари. Аномалию обнаруживают сразу после появления ребенка на свет или несколько позже — в пубертатном или зрелом возрасте. Это зависит от типа синдрома. У взрослых пациентов недуг чаще всего становится неожиданной находкой. Средний возраст больных — 25-40 лет.
Симптоматика патологии также определяется типом мальформации. Хоть синдром и считается врожденным пороком, не всегда его клинические признаки возникают с самого рождения. Иногда они обнаруживаются после 40 лет. У больных возникает цефалгия, напряженность мышц шеи, головокружение, нистагм, обмороки, атаксия, нарушения речи, парез гортани, тугоухость, падение остроты зрения, нарушение процесса глотания, стридор, парестезии, слабость мышц. У большинства пациентов симптомы заболевания полностью отсутствуют. Его обнаруживают случайно, во время комплексного диагностического обследования, проводимого совсем по другому поводу. Пациентам с бессимптомными формами не требуется лечение.
При постановке диагноза учитывают данные осмотра больного и его неврологического статуса, а также результаты томографического исследования мозга. Магнитно-ядерный резонанс позволяет точно и быстро определить наличие трансформации, уровень поражения и степень ущемления головного мозга. Лечение синдрома медикаментозное, физиотерапевтическое и оперативное. Больным выполняют операции по шунтированию мозга и декомпрессии краниовертебральной зоны.
Этиология
Причины синдрома в настоящее время точно не определены. Существует несколько теорий происхождения патологии, но до конца ни одна из них не имеет официального подтверждения. Мнения ученых-неврологов всего мира до сих пор расходятся.
Большинство медиков признают синдром врожденным недугом, сформированным в процессе эмбриогенеза под воздействием негативных факторов внешней среды, оказывающих свое пагубное действие на женский организм при беременности. К ним относятся: самостоятельные использование лекарств, алкоголизм, табакокурение, вирусные инфекции, ионизирующее излучение.

Врожденные причины:
- Дистопия мозжечковых миндалин за пределы задней черепной ямки, имеющей относительно малые размеры;
- Выталкивание растущих структур мозга через затылочное отверстие;
- Неправильное формирование в процессе эмбриогенеза и атипичное развитие в постнатальном периоде костной ткани, приводящее к деформации черепной коробки.
Некоторые ученые определенную роль в развитии синдрома отводят генетическому фактору. В настоящее время можно точно утверждать, что болезнь не связана с хромосомными аномалиями.
Другие ученые придерживаются иной точки зрения относительно происхождения синдрома. Они считают его приобретенным и объясняют свое мнение появлением симптомов патологии у взрослых лиц. Приобретенный синдром возникает под действием экзогенных факторов. У больного новорожденного ребенка череп может иметь нормальное строение без костных аномалий и гипоплазии.
Приобретенные причины:
- Родовой травматизм с поражением черепа и мозга,
- Воздействие цереброспинальной жидкости на стенки спинного мозга,
- Любые ЧМТ,
- Бурный рост мозга в условиях медленно растущих костей черепа.
Симптомы патологии долгое время отсутствуют у больных, а затем внезапно появляются под воздействием провоцирующих факторов: вирусов, травм головы, стрессов.
Опущение основных мозговых структур до шейных позвонков блокирует процесс перетекания ликвора из подпаутинного пространства в спинномозговой канал. Это приводит к дисциркуляторным изменениям. Ликвор, продолжая синтезироваться и никуда не оттекая, скапливается в головном мозге.
Ученый Киари в 1891 году выделил четыре типа аномалии:
- I – выход структурных элементов головного мозга за пределы задней черепной ямки, обусловленный недоразвитием костной ткани этой области. Этот тип клинически проявляется у лиц зрелого возраста.

выход структур мозжечка за пределы ЗЧЯ при аномалия 1 типа
- II — нарушения эмбриогенеза, приводящие к расположению структур мозжечка и продолговатого мозга ниже большого затылочного отверстия.

синдром Арнольда-Киари II типа
- III — эктопия мозговых структур в каудальном направлении с образованием энцефаломенингоцеле.

аномалия III типа
- IV — недоразвитый мозжечок не смещается и не выходит за пределы черепа. Поскольку отсутствует грыжевое выпячивание мозга, этот тип синдрома отсутствует в современной классификации.
Существуют два новых типа синдрома. Тип — мозжечок располагается достаточно низко, но при этом находится в черепной коробке. Тип 1.5 – промежуточная форма, сочетающая признаки I и II типов.
Выделяют три степени тяжести патологии:
- Первая — относительно легкая форма патологии без аномалий мозговых структур и характерных клинических проявлений.
- Вторая — наличие пороков развития ЦНС с врожденным недоразвитием головного мозга и подкорки.
- Третья — аномалии строения головного мозга со смещением мягких тканей в отношении твердых структур, образованием ликворных кист и сглаженностью извилин.
Симптоматика

Аномалия Киари I типа — самая распространенная форма синдрома, клинические признаки которой условно объединены в пять синдромов:
- Гипертензионный синдром проявляется цефалгией, подъемом артериального давления в утренние часы, напряженностью и гипертонусом шейных мышц, дискомфортом и болезненными ощущениями в шейном отделе позвоночника, диспепсическими явлениями, общей астенизацией организма У новорожденных детей возникает общее беспокойство, рвота фонтаном, тремор подбородка и конечностей, нарушается сон. Ребенок постоянно плачет, отказывается от груди.
- При наличии мозжечковых нарушений у больных изменяется произношение, речь становится скандированной, возникает вертикальный нистагм. Они жалуются на частые головокружения, рассогласованность движений, шаткость походки, дрожание рук, нарушение равновесия, дезориентацию в пространстве. Больные с большим трудом выполняют простые целенаправленные действия, в движениях отсутствует четкость и скоординированность.
- Поражение черепно-мозговых нервов проявляется признаками корешкового синдрома. У пациентов ограничивается подвижность языка и мягкого неба, что приводит к нарушению речи и проглатывания пищи. Их голос изменяется в сторону гнусавости и осиплости, речь становится неясной, дыхание затрудненным. Нарушение ночного дыхания отмечаются у большинства больных. У них возникает гипопноэ, центральное или обструктивное апное, при прогрессировании которого развивается острая дыхательная недостаточность. Лица с синдромом плохо слышат и видят, у них двоится в глазах и шумит в ушах. Со стороны органов зрения пациенты отмечают наличие светобоязни и боль при движении глазными яблоками. Офтальмологи часто обнаруживают анизокорию, спазм аккомодации или скотомы. Одним из основных симптомов синдрома является гипестезия – снижение чувствительности кожи лица и конечностей. Подобные патологическое изменения связаны с приглушенным реагированием рецепторов кожи на внешние раздражители: тепло или холод, уколы, удары. В тяжелых случаях нервное окончания вообще перестают воспринимать различные экзогенные воздействия.
- Сирингомиелический синдром — сложный симптомокомплекс, проявляющийся парестезией или онемением конечностей; изменением тонуса мышц и их гипотрофией, приводящей к миастеническим расстройствам; поражением периферических нервов, проявляющимся болью в конечностях; дисфункцией органов таза в виде затрудненной дефекации или самопроизвольного мочеиспускания; возможны артропатии — поражения суставов.
- У больных с пирамидальной недостаточностью снижается сила в нижних конечностях и способность к тонким движениям, ограничивается объем движений, повышается мышечный тонус — так называемая спастичность, например, спастическая походка. Повышение сухожильных рефлексов сочетается с одновременным снижением кожных рефлексов — брюшных. Возможно появление патологических рефлексов. У пациентов страдает мелкая моторика рук.
Любое неосторожное движение усиливает симптомы патологии, делает их более выраженными и яркими. Изменение положения головы — частая причина потери сознания.
Синдром Киари II типа имеет сходные клинические проявления. У новорожденных возникает паралич гортани, врожденный стридор, ночное апноэ, дисфагия, срыгивания, нистагм, гипертонус мышц рук, цианоз кожи. Аномалии III и IV типов не совместимы с жизнью.
Диагностические мероприятия

Аномалия Арнольда-Киари на снимке МРТ
Врачи-неврологи и невропатологи осматривают пациента и выявляют характерные особенности походки, изменение рефлексов и чувствительности на определенных участках тела, слабость в руках и прочие признаки. Все проявления мозжечкового, гидроцефального, бульбарного и прочих синдромов в совокупности позволяют врачу заподозрить аномалию.
После определения неврологического статуса больного требуется проведение комплексного неврологического обследования, включающего инструментальные методы — электроэнцефалографию, УЗИ головного мозга, реоэнцефалографию, ангиографию, рентгенографию. Эти методики выявляют лишь косвенные признаки патологии — изменения, происходящие в организме больного.
Ядерный магнитный резонанс лежит в основе особого нерентгенологического метода исследования – томографии. Этот спектроскопический анализ безопасен для большинства людей. Он дает изображение, состоящее из тонких срезов от магнитнорезонансного сигнала, проходящего через тело человека. На сегодняшний день именно МРТ позволяет быстро и точно поставить диагноз. Томография визуализирует структуру костей и мягкие ткани черепа, определяет пороки мозга и его сосудов.
Лечебный процесс
Лечение аномалии Киари комплексное, включающее медикаментозное воздействие, физиотерапевтические процедуры и хирургическое вмешательство. Именно оно в большинстве случаев помогает справиться с недугом и восстановить нормальную работу всего организма. Возможно применение средств народной медицины, которые дополняют, но не заменяют основное лечение. Использование фитосборов, отваров и настоев лекарственных трав должно быть одобрено лечащим врачом.
Лекарственная терапия и физиотерапия
Если больные испытывают сильную головную боль, боль в шее, мышцах и суставах, им назначают следующие группы препаратов:
- Обезболивающие средства – «Кеторол», «Пенталгин», «Анальгин».
- НПВС для уменьшения боли – «Мелоксикам», «Ибупрофен», «Вольтарен».
- Миорелаксанты для снятия напряжения с мышц шеи – «Мидокалм», «Сирдалуд».
Патогенетическое лечение синдрома включает:

- Препараты, улучшающие мозговое кровообращение – «Пирацетам», «Винпоцетин», «Циннаризин».
- Диуретики для уменьшения образования ликвора и с целью дегидратации – «Фуросемид», «Маннитол».
- Витамины группы В, поддерживающие работу нервной системы на оптимальном уровне и оказывающие антиоксидантное действие – «Тиамин», «Пиридоксин». Наиболее распространенные витаминные средства «Мильгамма», «Нейромультивит», «Комбилипен».
Если состояние больного признают крайне тяжелым, то его госпитализируют сразу в реанимационное отделение. Там пациента подключают к аппарату ИВЛ, устраняют имеющийся отек мозга, предупреждают инфекционные патологии и корректируют неврологические нарушения.
Физиотерапевтическое воздействие дополняет медикаментозное лечение, позволяет быстрее добиться положительных результатов, ускоряет процессы восстановления функций организма и выздоровления больных. Неврологи назначают:
- Криотерапию, оказывающую обезболивающий эффект, стимулирующую работоспособность желез внутренней секреции и укрепляющую иммунитет.
- Лечение лазером, улучшающее трофику и микроциркуляцию в очаге поражения.
- Магнитотерапию, оказывающую общее оздоравливающее действие и запускающую внутренние резервы организма.
В настоящее время особой популярностью пользуется кинезиологическая терапия, которая направлена на развитие умственных способностей и достижение физического здоровья через двигательные упражнения. Ее также включают в схему лечения данного синдрома.
Лечение не проводится вообще, если патология была обнаружена случайно, во время прохождения томографического обследования совсем по другому поводу, и у больного отсутствуют какие-либо характерные симптомы. За состоянием таких пациентов специалисты ведут динамическое наблюдение.
Оперативное вмешательство
Стойкие неврологические нарушения с парестезиями, дистонией мышц, параличами и парезами требуют проведения хирургической коррекции. Оперативное вмешательство показано также в тех случаях, когда медикаментозная терапия на дает положительного результата. Операции преследуют одну цель — устранение сдавления и ущемления мозга, а также восстановление нормальной циркуляции ликвора.

В настоящее время нейрохирурги спасают жизнь больным путем выполнения декомпрессивных и шунтирующих операций. В первом случае выпиливают часть затылочной кости с целью расширения большого отверстия, а во втором создают обходной путь для оттока ликвора по имплантационным трубкам с целью снижения его объема и нормализации внутричерепного давления.
После хирургического вмешательства всем пациентам показаны реабилитационные мероприятия. Если лечение было успешным, у больных восстанавливаются утраченные функции — дыхательные, двигательные, сердечно-сосудистые, нервные. В течение трех лет возможно рецидивирование патологии. В таких случаях больных признают инвалидами.
Видео: об операции при синдроме Арнольда-Киари
Народная медицина
Народные средства, применяемые при данной патологии, устраняют боль и расслабляют напряженные мышцы. Они эффективно дополняют традиционную терапию синдрома.
Наиболее популярные средства:
- Настой алтея для постановки компрессов,
- Прогревания пораженного места горячим куриным яйцом,
- Медовые компрессы,
- отвар папоротника или малины для приема внутрь.
Синдром Арнольда-Киари – порок развития, протекающий в бессимптомной форме или проявляющийся клинически с момента рождения. Патология имеет весьма разнообразную симптоматику и подтверждается с помощью МРТ. Лечебный подход к каждому пациенту индивидуален. Тактика лечения варьируется от симптоматического воздействия медикаментами до оперативного вмешательства с трепанацией черепа и удалением части мозговых структур.
Профилактика
Поскольку этиология синдрома окончательно не выяснена и нет конкретной информации о его патогенезе, предупредить развитие патологии не представляется возможным. Будущим родителям необходимо знать все о ведении здорового образа жизни и при планировании беременности стараться соблюдать указанные правила:
- Отказаться от пагубных привычек в виде табакокурения и употребления спиртных напитков,
- Обогащать свой рацион белковыми продуктами, фруктами, овощами, ягодами, исключив из него сладости и вредности,
- Своевременно обращаться к врачам за медицинской помощью,
- Принимать лекарственных препараты по назначению врача и в строго указанных дозировках,
- С профилактической целью принимать поливитамины,
- Беречь свое здоровье и наслаждаться жизнью.
Прогноз патологии неоднозначный. Консервативное лечение часто не дает положительных результатов. Хирургическое вмешательство, выполненное своевременно и в полном объеме, не всегда восстанавливает утраченные функции организма. Согласно статистическим данным эффективность такого лечения редко превышает 50-60%. Синдром третьей степени имеет неблагоприятный прогноз, поскольку поражаются многие мозговые структуры. При этом возникают несовместимые с жизнью функциональные нарушения.
Видео: лекция по синдрому Арнольда-Киали
Источник
Общая информация
Синдром Арнольда-Киари представляет собой набор признаков и симптомов, вызванных редкой мальформацией (отклонение от нормального развития, аномалия) задней черепной ямки; у пострадавших эта структура развита слабо, поэтому мозжечок выходит (выступает) из своего естественного участка через затылочное отверстие, расположенное у основания черепа.
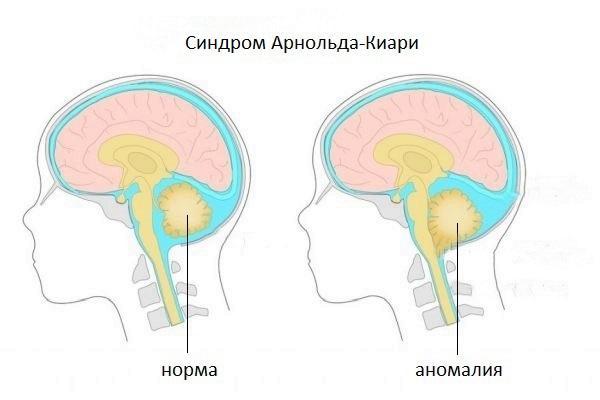
Есть четыре различных типа синдрома Арнольда-Киари; особенность, отличающая один тип от другого, является степень выпячивания, следовательно, доля вовлеченного материала мозжечка. Тип I является наименее тяжелым (иногда остается бессимптомным на протяжении всей жизни), тогда как IV тип наиболее тяжелый; однако уже со второго типа качество жизни больного ставится под угрозу.
Симптомы, характеризующие аномалии Арнольда-Киари многочисленны и варьируются от головных болей до слабости мышц и проч.
На сегодняшний день не существует лекарств, позволяющих устранить порок развития мозжечка, однако существуют способы лечения, позволяющие частично смягчить симптомы.
Что такое синдром Арнольда-Киари?
Синдром Арнольда-Киари, или мальформация Арнольда-Киари — структурное изменение мозжечка, характеризующееся его смещением вниз, именно в направлении позвоночного канала и затылочного отверстия, базальные части полушария мозжечка.
Простыми словами, это грыжа мозжечка, при которой часть мозжечка выступает из затылочного отверстия, проникая в позвоночный канал.
Синдром Арнольда-Киари получил свое название от двух врачей, которые впервые описали его, Арнольда Джулиуса и Ганса Киари.
Причины и факторы риска
Исследователи полагают, что синдром Арнольда-Киари может иметь наследственное происхождение, так как обнаруживалась среди членов одной семьи. Тем не менее, генетические условия, вызывающие заболевание (т.е., какие и сколько генов участвуют) и тип передачи еще предстоит выяснить.
Исходя из серьезности выпячивания и момента жизни, в котором он возникает, заболевание можно разделить на 4 различных типа, идентифицированных первыми четырьмя римскими числами (I, II, III и IV).
Первые два типа по сравнению со вторыми более распространены и менее серьезны; Тип III и тип IV, на самом деле, очень редки и несовместимы с жизнью.
— Мальформация I типа.
Первая степень синдрома протекает бессимптомно (т.е. без явных симптомов), по крайней мере, до конца детства или юности.
Причина его возникновения кроется в уменьшенном черепном пространстве: в таких условиях часть мозжечка (именно миндалина(и), расположенная(ые) с нижней стороны), из-за недостатка места, вынуждена проникать в затылочное отверстие и входить в позвоночный канал.
Примечание: у некоторых взрослых людей с синдром Арнольда-Киари 1 типа все в порядке и они ведут совершенно нормальную жизнь. Это связано с тем, что аномалия мозжечка не настолько серьезна, чтобы вызывать симптомы или нарушения. Поэтому очень часто эти субъекты игнорируют свое состояние или узнают о нем по чистой случайности.
— Мальформация II типа.
2 тип мальформации Арнольд-Киари является врожденным заболеванием, которое присутствует с рождения ребенка, и всегда протекает симптоматически.
По сравнению с 1 степенью он характеризуется большим выпячиванием черепной ямки, при котором помимо миндалин мозжечка также выпячивает часть мозжечка (называемая червь мозжечка) и венозный сосуд.
Почти всегда мальформация Арнольд-Киари II типа ассоциируется с особой формой расщелины позвоночника, называемой миеломенингоцеле.
Среди различных последствий этой аномалии выделают: блокирование потока ликвора (спинномозговой жидкости) через затылочное отверстие (что приводит к состоянию, называемому гидроцефалией) и прерывание нервных сигналов.
Первоначально термин Арнольд-Киари относился только ко 2 типу заболевания. Теперь, её обычно используют для всех форм болезни.
— Мальформация III типа.
Присутствующий с рождения, III тип порока вызывает серьезные неврологические проблемы, настолько, что часто несовместимы с жизнью. В этих случаях на самом деле наблюдается выпячивание мозжечка, и по этой причине говорится о затылочном энцефалоцеле.
Обычно III тип состояния характеризуется гидроцефалией и сирингомиелией; последний представляет собой особое состояние, характеризующееся наличием одной или нескольких кист в позвоночном канале.
— Мальформация IV типа.
Мальформация Арнольда-Киари IV типа характеризуется отсутствием развития части мозжечка (недоразвитие мозжечка).
Аномалия врожденная и абсолютно несовместима с жизнью.
Связанные расстройства
Врачи и ученые отметили, что следующие заболевания являются частыми среди людей с мальформацией Киари:
- гидроцефалия;
- сирингомиелия;
- сколиоз;
- синдром Марфана;
- синдром Элерса-Данлоса.
Эпидемиология
Точная частота пороков развития неизвестна; это связано с тем, что у некоторых даже взрослых людей с I типом мальформации Арнольда-Киари никаких симптомов нет, и они кажутся совершенно нормальными (поэтому болезнь недиагностируется).
Несколько достоверных эпидемиологических исследований сообщают, что:
- I тип симптоматичен у 1 из 100 детей;
- II тип особенно широко распространен в популяциях кельтского происхождения;
- женщины страдают в 3 раза чаще, чем мужчины.
Симптомы и осложнения

4 типа заболевания имеют разные симптомы и признаки.
Ниже приводится таблица с точным описанием симптомов, которые характеризуют I, II и III типы синдрома.
Для IV типа невозможно проследить симптоматику, так как это состояние неизбежно и внезапно приводит к гибели плода.
| Мальформация I типа | Мальформация II типа | Мальформация III типа |
Когда у больного 1 стадия, симптомы следующие:
| Синдром Арнольда-Киари II типа характеризуется теми же симптомами, что и тип I, с той разницей, что они имеют более выраженную интенсивность и присутствуют всегда. Кроме того, если он сопровождается миеломенингоцеле (см. ниже), II тип состояния также вызывает:
| Люди с мальформацией III типа страдают от серьезных неврологических проблем (часто несовместимых с нормальной жизнью), гидроцефалии и сирингомиелии. Последний, характеризуется образованием одной или нескольких кист внутри спинного мозга, и может вызывать:
|
Расщелина позвоночника (миеломенингоцеле).
Расщелина позвоночника — врожденный порок развития позвоночника, из-за которого менинги, а иногда и спинной мозг выходят из своего места (обычно они ограничены позвонками). Миеломенингоцеле является наиболее тяжелой формой расщелины позвоночника: у пораженных выпячивают менинги и спинной мозг из позвоночной камеры и образуют мешок на уровне спины. Эта сумка, хотя и защищена слоем кожи, подвержена внешним воздействиям и постоянно подвергается риску серьезных, а в некоторых случаях даже смертельных инфекций.
Синдром Арнольда-Киари типа II, III и IV видны уже в пренатальном возрасте (т.е., когда пораженный ребенок все еще находится в материнской утробе) при ультразвуковом исследовании.
Что касается I типа, желательно обратиться к врачу, как только появятся типичные симптомы, о которых говорилось выше. Важно также проходить своевременные обследования, так как в результате последних могут возникнуть другие сопутствующие нарушения.
Осложнения
Осложнения синдром Арнольда-Киари связаны с ухудшением выпячивания мозжечка или патологическими состояниями, связанными, следовательно, с гидроцефалией, миеломенингоцеле, сирингомиелией и проч.
Ухудшение выпячивания (протрузии), обусловленное повышенным давлением черепа на мозжечок, очевидно, предполагает обострение симптомов.
Диагностика
Диагностические тесты, которые позволяют установить степень протрузии мозжечка через затылочное отверстие (таким образом, устанавливая тип мальформации Арнольда-Киари):
- Магнитно-резонансная томография (МРТ). Благодаря формированию магнитных полей, он позволяет получить детальное изображение мозжечка и позвоночного канала, не подвергая пациента вредному ионизирующему излучению.
- Компьютерная томография (КТ) дает четкие изображения внутренних органов, в том числе мозжечка и спинного мозга. Во время его выполнения субъект подвергается минимальному воздействию вредного ионизирующего излучения.
КТ и МРТ, которым предшествует точное физическое обследование, имеют основополагающее значение для выявления любых патологий, связанных с синдромом Арнольда-Киари.
Таблица. Как и когда диагностируется аномалия Арнольда-Киари развития заболевания.
| Тип порока развития | Когда и как это можно диагностировать? |
| I | В позднем детстве или позднем подростковом возрасте посредством объективных обследований с последующим КТ и/МРТ. |
| II | В дородовом возрасте при УЗИ. При рождении и в раннем детстве, посредством объективных обследований, компьютерной томографией и/или МРТ. |
| III | В дородовом возрасте при УЗИ. После рождения и в раннем детстве, посредством объективных обследований, компьютерной томографией и/или МРТ. |
| IV | В дородовом возрасте при УЗИ. |
Лечение
Синдром Арнольда-Киари неизлечим. Однако существуют как фармакологические, так и хирургические методы терапии, позволяющие частично смягчить признаки заболевания.
— Медикаментозная терапия.
Пациенты с мальформацией Арнольда-Киари I типа, страдающий от головной боли и боли в шее и/или лице, могут принимать обезболивающие препараты.
Выбор наиболее подходящих лекарств для конкретного случая остается за лечащим врачом.
— Хирургическое лечение.
Цель хирургического лечения состоит в том, чтобы уменьшить давление, оказываемое черепом, чтобы предотвратить повреждение мозжечка и спинного мозга.
Для достижения этой цели есть несколько процедур, таких как:
- Декомпрессия задней черепной ямки, во время которой хирург удаляет часть задней части затылочной кости.
- Декомпрессия спинного мозга с помощью ламинэктомии (декомпрессионная ламинэктомия). Во время его выполнения хирург удаляет пластинку второго и третьего шейного позвонка. Пластинка — это позвоночная часть, отделяющая отверстие, через которое проходит спинной мозг..
Примечание: иногда декомпрессия задней ямки и декомпрессивная ламинэктомия выполняются одновременно. - Декомпрессионный разрез твердой мозговой оболочки. При разрезе твердой мозговой оболочки или наружного менинга пространство, доступное мозжечку, увеличивается, а давление на его повреждение уменьшается. Чтобы покрыть и защитить трещину, созданную разрезом, хирург пришивает на нее кусок искусственной ткани (или взятый из другой части тела).
- Хирургическое шунтирование (создание дополнительного пути в обход пораженного участка). Это, по сути, дренажная система, состоящая из гибкой трубки, позволяющей удалять спинномозговую жидкость, в случае гидроцефалии, или опорожнять цисту(ы), в случае сирингомиелии. Не исключено, что пациенты с гидроцефалией будут вынуждены проходить хирургическим шунтом всю жизнь.
— Осложнения хирургического вмешательства.
Риски, связанные с хирургией, различны. Фактически возможно появление:
- кровотечений;
- повреждение структур головного мозга и/или спинного мозга;
- инфекционный менингит;
- проблемы с заживлением ран;
- необычные скопления жидкости вокруг мозжечка.
Помните, что любое повреждение головного или спинного мозга, произошедшее во время операции, непоправимо. Поэтому, прежде чем подвергнуться больного вмешательству любого типа, лечащий врач выявит любые риски и осложнения необходимой процедуры.
Прогноз
Синдром Арнольда-Киари типа II, III и IV никогда не имеют положительного прогноза, поскольку, помимо того, что неизлечимы, могут вызывать серьезные неврологические нарушения или даже быть несовместимыми с жизнью.
Прогноз для пациентов с I типом часто неизвестен. Многие люди с этим заболеванием не имеют никаких симптомов, и невозможно предсказать, будут ли симптомы развиваться в будущем. Другие люди с мальформацией Арнольда-Киари могут испытывать головокружение, мышечную слабость, онемение, проблемы со зрением, головную боль или проблемы с равновесием и координацией. У этих людей не всегда возможно предсказать, будут ли симптомы ухудшаться с течением времени.
Людям с пороком 1 типа важно регулярно проходить медицинские обследования, чтобы быть под наблюдением врача при появлении любых новых симптомов.
Источник