Микроделеционные синдромы когда проявляются у ребенка

Группа хромосомных патологий, называемых «синдромы микроделеции 22q11.2», – одна из причин возникновения умственной отсталости у детей. Такие нарушения не всегда удается зафиксировать методами цитогенетической диагностики, но можно выявить с помощью ультразвукового исследования.

Сергей Адольфович, что представляют собой микроделеционные синдромы?
Микроделеционные синдромы (МДС) – это особый вид хромосомных заболеваний, при которых происходит потеря микроскопического участка хромосомного материала. Стоит особо остановиться на микроделеции участка q11.2 хромосомы 22, которая является одной из самых распространенных у человека. Задача выявления данной группы синдромов достаточно сложна из-за различных форм проявления заболевания. Клинически синдром может проявляться судорогами, возможны затруднения при вскармливании, новорожденные имеют повышенный риск внезапной смерти. Наиболее же частыми причинами смерти детей с этим синдромом в раннем возрасте являются тяжелые врожденные пороки сердца и инфекции, а выжившие дети часто отстают в физическом и умственном развитии.
Какова частота возникновения таких патологий?
Эта аномалия встречается с относительно высокой частотой – 1 случай на 4000–7000 живорожденных. Это второй по частоте синдром после синдрома Дауна. Но если для последнего фактором риска рождения больного ребенка является возраст матери, то МДС 22q11.2 не зависит от возраста женщины. И совсем молодые женщины (до 30 лет) могут быть подвержены риску рождения детей с подобными отклонениями, при этом очень большое количество случаев остается не диагностированным ни во время беременности, ни даже после рождения ребенка.
Чем опасны микроделеционные синдромы?
Подобные хромосомные нарушения могут развиваться с минимальными проявлениями. К примеру, если во время скринингового исследования обнаруживается изолированный порок сердца (кстати, такие патологии диагностируются с помощью УЗИ уже в первом триместре – со срока 11 недель и 2 дня), беременную женщину в дальнейшем, как правило, направляют к специалисту-кардиологу для определения хирургической тактики. Другие же проявления микроделеционных синдромов, которые поражают не только сердечно-сосудистую, но и дыхательную, иммунную системы и влияют на умственное развитие ребенка, могут остаться незамеченными. Причем некоторые симптомы начинают проявляться в возрасте около 5 лет, когда выясняется, что ребенок, которого нужно готовить к школе, необучаемый и страдает умственной отсталостью. А между тем, чем позже ставится точный диагноз, тем тяжелее последствия МДС для ребенка, так как могут формироваться физические отклонения, опасные для жизни, задержка развития и проблемы в школе.

Как этого избежать?
Врач-диагност, выявляя определенные пороки развития сердца плода, обязательно должен рассматривать все признаки и прежде всего обратить внимание на развитие тимуса (вилочковой железы). Здесь важны не только знания, но и большой опыт вместе с мануальными навыками и качеством ультразвукового оборудования. Возможности, предоставляемые клиникой, позволяют мне совершенствоваться в этом направлении.
Есть ли другие маркеры МДС?
Опасность микроделеций состоит еще и в том, что только 75–80% развивающихся синдромов сопровождаются ярко выраженными пороками сердца. Остальные имеют менее заметные проявления, которые также обязательно нужно отслеживать и учитывать уже в первом триместре, и тут важную роль играют лицевые аномалии. В условиях нашей клиники мне удалось получить патент на изобретение способа ранней диагностики лицевых аномалий и связанных с ними синдромов.
Что нужно делать будущей матери во время беременности, чтобы родить здорового ребенка?
Для начала необходимо исключить все возможные осложнения. Нужно провести подробную и качественную диагностику первого триместра, оценить принадлежность к группе риска, особенно если предыдущие беременности окончились неудачно, так как более 1/4 случаев заболевания являются семейными. МДС 22q11.2 не является исключительной редкостью, и долю больных с этим синдромом можно оценить как 5–10% среди всех детей, нуждающихся в оперативном лечении врожденного порока сердца, и 5% среди детей, имеющих расщелину нёба, не сочетающуюся с расщелиной губы. Огромную роль играет грамотная консультация семьи генетиком с использованием новейших знаний и возможностей лабораторной диагностики.
Дата публикации: 12.09.16
Источник

УЗИ аппарат HM70A
Универсальный переносной УЗ-аппарат экспертного класса по доступной цене. Монокристальные датчики, полноэкранный режим отображения, эластография, 3D/4D в корпусе ноутбука. Гибкая трансформация в стационарный сканер при наличии тележки.
Пренатальная диагностика — раздел современной медицины, находящийся на стыке социально лабораторных технологий переводят возможности пренатальной диагностики на качественно новый уровень. Суть медико-генетического пренатального консультирования заключается в определении прогноза для жизни и здоровья плода с наследственной и врожденной патологией, в объяснении вероятности неблагоприятного исхода беременности и помощи семье в принятии решения о деторождении при неблагоприятном прогнозе не только для жизни, но и для здоровья будущего ребенка [1].
Микроделеционные синдромы (МДС) — особый вид хромосомных заболеваний, при котором происходит потеря участка хромосомного материала, что не удается зафиксировать рутинными стандартными методами цитогенетической диагностики [2]. Микроделеция может быть следствием разрыва хромосомы или результатом неравного кроссинговера. Формально микроделеции относят к хромосомным аномалиям, поскольку они меняют количество генов, а не их структуру, но с практической точки зрения они наследуются как аутосомно-доминантные моногенные заболевания. Большинство микроделеций возникает в половых клетках здоровых родителей (до 70% всех МДС), и тогда риск повторного возникновения данной аномалии в семье минимален. Если же один из родителей болен (в 30% всех встречающихся МДС), то риск повторного рождения больного ребенка для этого родителя составляет 50%. В литературе сообщалось о возможном наследовании микроделеций от внешне здоровых родителей, однако в подавляющем большинстве случаев, если у родителя есть микроделеция, то она в той или иной степени проявляется клинически [3].
Можно отметить, что клиническая картина большинства МДС имеет классические черты частых хромосомных заболеваний, включая умственную отсталость различной степени, комплекс дизморфических изменений лица и некоторых пороков развития [4]. Некоторые из известных МДС у плодов проявляются как ультразвуковые маркеры частых хромосомных болезней (врожденные нарушения строения органов, изменения/особенности фенотипа, лицевые дизморфии) и поэтому могут быть заподозрены в ходе проведения пренатального ультразвукового исследования в разные сроки беременности.
Возможности лабораторной диагностики МДС при исследовании плодного материала вошли в рутинную практику врачей, занимающихся пренатальной диагностикой на современном уровне. Этот вид хромосомных аномалий возможно выявить при помощи специальных зондов на конкретный участок хромосомы при помощи флуоресцентного неизотопного варианта гибридизации (FISH). Однако ограничения этого метода заключаются в том, что выбор зонда должен быть строго таргетным, прицельным: на конкретный участок конкретной хромосомы, что весьма ограничивает клиническое применение метода, так как очень многие МДС по клиническим характеристикам схожи и могут выглядеть фенотипически идентично. В практике наиболее часто применяется хромосомный микроматричный анализ (ХММА) — это тест для определения структурных изменений ДНК, при которых происходит изменение количества генетического материала в виде его нехватки (делеции) или избытка (дупликации), используя специальные геномные микрочипы [5].
Клиническое наблюдение
Беременная 30 лет обратилась в медико-генетическое отделение Московского областного НИИ акушерства и гинекологии в связи с выявлением в окружном кабинете пренатальной диагностики по месту жительства врожденной аномалии развития обеих стоп у плода. Беременность первая, брак первый, не родственный. Супруги профессиональных вредностей не имеют. По результатам скрининга I триместра риск хромосомных аномалий низкий. Эхография проводилась на сканере WS80A (Samsung Medison, Ю. Корея). На момент осмотра гестационный срок беременности 21 нед. При эхографии размеры плода соответствовали гестационному сроку. Были выявлены множественные особенности строения плода: аномальная установка обеих стоп с сохраненной их двигательной активностью, увеличение шейной складки до 8 мм, расширение большой цистерны головного мозга до 10 мм (рис. 1, 2). Проведено медико-генетическое консультирование, предварительный прогноз для жизни плода определен как условно благоприятный, однако по комплексу выявленных множественных особенностей развития плода рекомендовано проведение инвазивной пренатальной диагностики. После согласия пациентки был проведен амниоцентез. Семья приняла решение о расширенном обследовании плодного материала с проведением хромосомного микроматричного тестирования. По результатам обследования была обнаружена микроделеция длинного плеча (q) 10-й хромосомы с позиции 81684402 до позиции 88847443, захватывающая регионы с 10q22.3 по 10q23.2. В заключении описаны множественны гены, расположенные в районе дисбаланса: SFTPD, PLAC9, ANXA11, MAT1A, DYDC1, NRG3, CDHR1, LRIT1, RGR, MIR346, WAPL, OPN4, LDB3, BMPR1A, MMRN2, SNCG, GLUD1. Данная микроделеция в литературе описана как патогенная. Область 10q22.3-q23.2 характеризуется сложным набором низкокопийных повторов (LCRs), которые могут приводить к различным геномным изменениям. Делеции хромосомы 10q22.3-q23.2, включая ген BMPR1A, были связаны с лицевыми дизморфиями, задержкой развития и множественными врожденными аномалиями [6, 7].
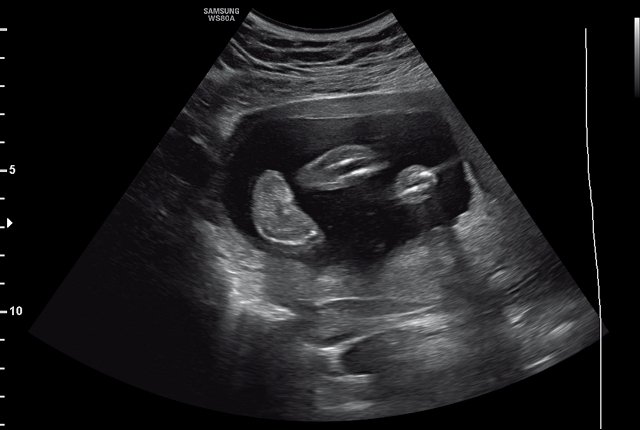
Рис. 1. Аномальная установка стопы у плода (беременность 21 нед).
Рис. 2. Увеличение шейной складки, расширение большой цистерны головного мозга (беременность 21 нед).
После получения результатов обследования плодного материала проведено повторное медико-генетическое консультирование с тщательным сбором анамнеза и с внимательным осмотром беременной. Обнаружены «мягкие» лицевые дизморфии: гипотелоризм, незначительная асимметрия лица, низко расположенные диспластичные ушные раковины, клинодактилия мизинцев (рис. 3, 4). При углубленном сборе анамнеза выявлено, что у пациентки в детстве отмечалась задержка речевого развития до 6 лет, а в 12 лет она перенесла ортопедическую операцию на обеих стопах в связи с аномальной их установкой (двусторонняя внутренняя косолапость). Однако стоит отметить, что на данный момент женщина имеет 2 высших образования, особенностей в поведенческих и коммуникативных реакциях не выявлено. В связи с выясненными данными анамнеза и осмотра беременной для определения происхождения микроделеции у плода и формулировки окончательного прогноза для жизни и здоровья будущего ребенка решено обследовать родителей. При проведении ХММА у беременной была обнаружена микроделеция длинного плеча (q) 10-й хромосомы с позиции 81646459 до позиции 89146603, захватывающая регионы с 10q22.3 по 10q23.2. Гены, расположенные в районе дисбаланса, такие же, как и у плода. Учитывая все полученные клинические и лабораторные данные, включающие сбор анамнеза и лабораторное подтверждение наследования МДС от матери, семья приняла решение пролонгировать данную беременность, однако в сроке 26 нед произошла антенатальная гибель плода.
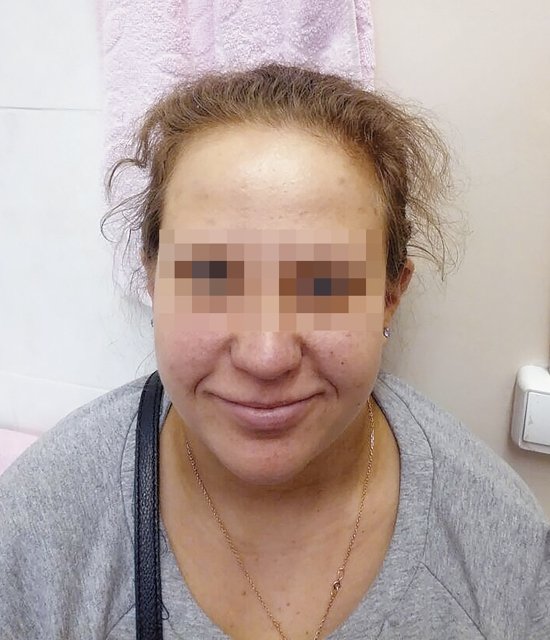
Рис. 3. Фенотип беременной с микроделеционным синдромом (анфас).
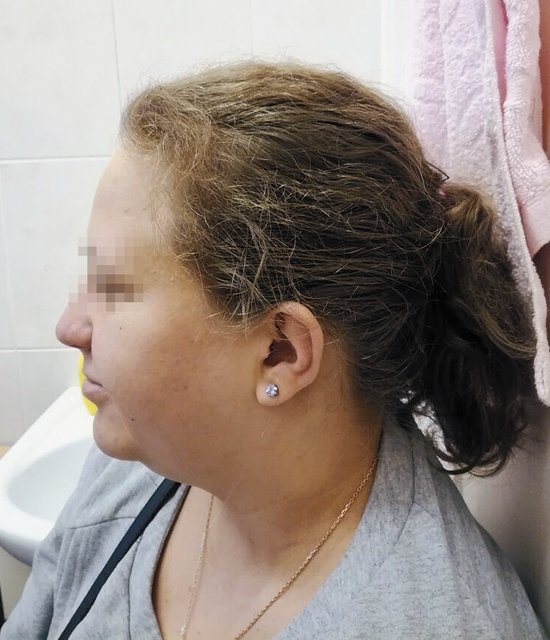
Рис. 4. Фенотип беременной с микроделеционным синдромом (профиль).
Заключение
В настоящее время, когда новые лабораторные методы исследования плодного материала все чаще входят в ежедневную практику врача пренатальной диагностики, нужно очень тщательно интерпретировать полученные данные. И если раньше пренатальная диагностика была реальной только для некоторых тяжелых инвалидизирующих заболеваний, то в современной диагностике доступны сотни наследственных и врожденных болезней с абсолютно разными прогнозами. Это порождает новые, ранее неизвестные сложности для медико-генетического консультирования как в отношении спектра болезней, подлежащих диагностике, так и при выработке врачебной тактики после диагностики с формулировкой адекватного прогноза для жизни и здоровья будущего ребенка. И если при ХММА установлены неоднозначные данные, необходимы обязательное дополнительное исследование супругов, тщательный сбор семейного анамнеза, осмотр с определением фенотипических особенностей родителей. Все эти мероприятия необходимы для решения вопроса о природе происхождения заболевания и корректировки возможных прогнозов для жизни и здоровья будущего ребенка.
Литература
- Баранов В.С., Кузнецова Т.В., Кащеева Т.К., Иващенко Т.Э. Пренатальная диагностика наследственных болезней. Состояние и перспективы. СПб., 2017: 14-100. 2. Lee C., Iafrate A.J., Brothman A.R. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders // Nat Genet. 2007; 39 (Suppl. 7): 48-54.
- Кеннет Л. Джонс. Наследственные синдромы по Д. Смиту. М.: Практика, 2011. 997 с.
- Manning M., Hudgins L. Professional Practice and Guidelines Committee «Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities» // Genet Med. 2010; 12: 742-745.
- Miller D.T., Adam M.P., Aradhya S. et al. Consen sus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with de velopmental disabilities or congenital ano ma lies // Am J Hum Genet. 2010; 86: 749-764.
- https://decipher.sanger.ac.uk
- https://www.omim.org/
УЗИ аппарат HM70A
Универсальный переносной УЗ-аппарат экспертного класса по доступной цене. Монокристальные датчики, полноэкранный режим отображения, эластография, 3D/4D в корпусе ноутбука. Гибкая трансформация в стационарный сканер при наличии тележки.
Источник
Генетическое исследование, позволяющее выявить причинный фактор – мутацию (отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15-й отцовской хромосоме (синдром Прадера — Вилли) или мутацию материнского генетического материала аналогичного генетического региона (синдром Ангельмана, а также мутация (выпадение одного из участков) 22-й хромосомы, ведущая к гипоплазии вилочковой и паращитовидной желез (синдром Ди Джорджи).
Синонимы русские
Тест на синдромы «смежных генов», FISH-тест на генетические аномалии, FISH-анализ на Прадера – Вилли, Ангельмана, Ди Джорджи.
Синонимы английские
The test for syndromes of «adjacent genes», FISH analysis on Prader-Willy syndrome (PWS), FISH analysis for Angelman syndrome (AS), FISH analysis for Di George syndrome (DGS), FISH-test for genetic abnormalities.
Метод исследования
Дифференциальное окрашивание хромосом.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Исследование проводится в состоянии сытости, не рекомендуется сдавать кровь на данное исследование натощак.
- Исключить (по согласованию с врачом) прием антибактериальных и химиотерапевтических препаратов в течение 14 дней до исследования.
- Исследование рекомендуется проводить не ранее чем через 2 недели после перенесенных инфекционных/острых воспалительных заболеваний.
Общая информация об исследовании
Цитогенетический анализ проводится методом флуоресцентной гибридизации in situ (FISH, от англ. fluorescence in-situ hybridization), который является одним из самых современных методов диагностики хромосомных аномалий. Подробнее с методом можно ознакомиться здесь (https://helix.ru/kb/item/12-052).
Одним из преимуществ FISH-теста является его способность определять микроделеции, которые не выявляются с помощью классического кариотипирования или полимеразной цепной реакцией (ПЦР). Это имеет особое значение при диагностике микроделеционных синдромов, например Прадера – Вилли, Ангельмана, Ди Джорджи. Делеции — это хромосомные перестройки, при которых происходит потеря участка хромосомы. Микроделеции часто характеризуются сложным клиническим и поведенческим фенотипом, возникающим в результате дисбаланса нормального количества генов, расположенных в этом конкретном сегменте хромосом.
Микроделеционные синдромы определяются как группа клинически узнаваемых расстройств, характеризующихся небольшой утратой хромосомного сегмента, охватывающего несколько соседних генов, каждый из которых потенциально может вносить свой вклад в фенотип. Эти синдромы называют еще синдромами «смежных генов». Большинство клинически значимых микроделеций, по-видимому, происходят спорадически, но в ряде случаев может обнаруживаться наследственная связь.
Велокардиофациальный синдром — наиболее частая известная интерстициальная делеция, обнаруженная у человека с частотой 1 из 4000 живорождений. Большинство потерь являются результатом события de novo, и только около 5-10% наследуются аутосомно-доминантным способом. Для диагностики этого синдрома используется несколько диагностических меток, включая синдром (аномалию) Ди Джорджи (DGS).
Синдром Ди Джорджи связан с делецией 22q11.2 и встречается у 1:3000-1:20000 новорождённых, с одинаковой частотой как у мальчиков, так и у девочек. Клинически он проявляется нарушениями иммунной системы, вплоть до тяжелого иммунодефицита из-за недоразвитости или отсутствия тимуса, гипопаратиреозом, пороками сердца, расщелиной неба, умственной отсталостью разной степени выраженности, катарактой (помутнение хрусталика). Также могут быть пороки развития мочевыделительной системы, проблемы с питанием. Больные, как правило, низкого роста и с характерными чертами лица (подбородок и рот маленькие, переносица расширена).
Синдром Прадера — Вилли (75% случаев) и 80% случаев возникновения синдрома Ангельмана связаны с микроделецией 15q11-13. Идентифицированы различные молекулярные механизмы, приводящие к этой потере. Примечательно, что при этом каждый из синдромов может сопровождаться и другими мелкими «молекулярными поломками».
Синдром Прадера-Вилли (PWS) представляет собой сложное мультисистемное расстройство, характеризующееся множеством клинических проявлений. Встречается патология у 1:12000-1:15000 новорождённых, которые в большинстве случаев рождаются преждевременно. Как правило, пациенты с данной аномалией небольшого роста, с короткими конечностями, имеют типичные черты лица: небольшой лоб, миндалевидной формы глаза, недоразвитую верхнюю челюсть, тонкую верхнюю губу и опущенные углы рта. С самого рождения наблюдается гиперфагия (неконтролируемое потребление пищи) и в результате — ожирение, тяжелая мышечная гипотония. Также характерны гипогонадизм (синдром, сопровождающийся недостаточностью функций половых желез и нарушением синтеза половых гормонов) с недостаточным ростом во время полового созревания, нарушения терморегуляции и дневная сонливость.
Синдром Прадера — Вилли рассматривается как многоэтапное расстройство, характеризующееся тремя различными фазами:
- Первая, «гипотоническая фаза», характеризуется разной степенью выраженности гипотонии во время неонатального периода и раннего младенчества, слабым криком, гипотермией, гипогенитализмом и слабым рефлексом сосания. В течение первого года дети с PWS дружелюбные, спокойные и ласковые.
- «Гиперфагическая фаза» обычно начинается в 1-2 года, характеризуется повышенным аппетитом, ранним началом детского ожирения, физической бездеятельностью, сниженной болевой чувствительностью, нарушенной терморегуляцией, психомоторной отсталостью, трудностью сочетанной речи и умственными нарушениями. Одновременно с изменением структуры питания меняется и поведение детей — оно становится социально неадекватным и неправильно эмоционально окрашенным (например, немотивированные вспышки гнева), появляются упрямство, лабильность настроения, импульсивность, беспокойство и обсессивно-компульсивные симптомы.
- Третья фаза — «подростковый возраст и взрослая жизнь». На этом этапе преобладают проблемы со здоровьем, связанные с ожирением (например, сколиоз, гипертония, гиперхолестеринемия, остеопороз и т.д.). Около 10% подростков и взрослых имеют серьезные психиатрические проблемы. Примечательно, что психотические эпизоды у пациентов с PWS имеют много общих черт, включая острое начало, полиморфную и колеблющуюся симптоматику с тревогами, агитацией, аномальными верованиями и слуховыми галлюцинациями. Эти эпизоды классифицируются как острый циклоидный психоз.
Синдром Ангельмана (AS) встречается примерно у 1:10000-30000 новорождённых, и с первых лет жизни наблюдается задержка умственного и неврологического развития. Способность к невербальному общению сохранена, больные понимают больше, чем способны выразить.
К типичным чертам при данной патологии относятся брахицефалия (короткоголовость), микроцефалия (недоразвитость черепа, маленькая голова), большой рот с широко расставленными зубами, нижнечелюстной прогнатизм (выпячивание нижней челюсти, затрагивающее более низкую треть лица), глубокие и голубые глаза и гипопигментация, практически постоянная улыбка. «Типичное лицо» становится очевидным в возрасте от одного года до четырех лет, и с увеличением возраста происходит огрубление и усиление характерных черт. У пациентов с AS наблюдается гипотония с гипертонией конечностей и высокий риск развития сколиоза. Все пациенты имеют умственную отсталость с различными речевыми расстройствами. С первых лет жизни отмечаются различные неврологические нарушения (например, дрожание уголков рта, выталкивание языка, немотивированное хлопанье руками, тики). У 80% детей в возрасте от 1 мес. до 5 лет встречаются эпилептические припадки. Их трудно контролировать, и они могут быть разнообразными, начиная от атипичного отсутствия судорог, тонико-клонических припадков, миоклонических приступов и тонических приступов до эпилептического статуса. Модели ЭЭГ, наблюдаемые у пациентов с синдромом Ангельмана, очень характерны и наблюдаются у пациентов с/без судорог и могут играть важную диагностическую роль в соответствующем клиническом контексте. Уже через 10 недель после рождения могут наблюдаться приступы легко спровоцированного продолжительного смеха. Дети, как правило, гиперактивны, имеют пониженную способность концентрировать внимание и различные нарушения сна. С возрастом походка становится медленная, атаксическая и жесткая с характерным положением поднятых рук с изгибами запястий и локтей (дерганная походка марионетки). Характерны нарушения координации, хаотичные движения рук и ног. Люди с AS плохо переносят душные помещения и повышенную температуру. Кроме классических проявлений синдрома, существуют разнообразные вариации, связанные с различными генетическими причинами. Например, более мягкая эпилепсия отмечена у пациентов со спонтанной мутацией материнской копии, которая вызывает отсутствие преобразования в мозге копии гена UBE3A.
FISH-анализ позволяет достоверно диагностировать общеизвестные микроделеционные синдромы и является стандартным диагностическим методом при данной патологии. Исследование позволяет установить этиологию заболевания и сделать прогноз по дальнейшему его течению.
Для чего используется исследование?
- Для диагностики микроделеций.
Когда назначается исследование?
- Пренатальная диагностика при подозрении на наличие отклонений в развитии плода.
- Постнатальная диагностика генетической патологии у ребенка при наличии соответствующих клинических признаков.
- При планировании последующих беременностей, если в семье есть ребенок с микроделеционным синдромом.
Что означают результаты?
Положительный результат говорит о наличии в исследуемом образце микроделеции.
Отрицательный результат указывает на отсутствие искомой микроделеции, но не может достоверно опровергать заболевание, так как синдром может быть вызван другим генетическим сбоем (например, однородительской дисомией, транслокацией родительских хромосом). В процентном соотношении вероятность этого значительно меньше, но все же возможна.
Синдром Прадера-Вилли:
- встречается у 1:25000- 1:10000 новорождённых;
- с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно.
Синдром Ангельмана:
- встречается примерно у 1:10000 новорождённых;
- с первых лет жизни наблюдается задержка умственного и неврологического развития.
Синдром Ди Джорджи:
- встречается у 1:3000-1:20000 новорождённых, в равной доле как у мальчиков, так и у девочек;
- характерна триада симптомов: гипоплазия тимуса и паращитовидной железы, врождённый порок сердца;
- 5-10% — аутосомно-доминантный тип наследования.
Важные замечания
FISH-тест часто используется совместно с другими методами молекулярной и цитогенетической диагностики.
Также рекомендуется
[40-006] Беременность — Пренатальный скрининг трисомий I триместра беременности (синдром Дауна)
[16-001] Исследование кариотипа
[40-007] Беременность — Пренатальный скрининг трисомий II триместра беременности
Кто назначает исследование?
Педиатр, врач-генетик, эндокринолог, невролог.
Литература
- Shaffer, L.G., Ledbetter, D.H., and Lupski, J.R. 2001, Molecular cytogenetics of contiguous gene syndromes: mechanisms and consequences of gene dosage imbalance. In: The Metabolic & Molecular Bases of Inherited Disease, C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle (Eds.), McGraw-Hill, Medical Publishing Division, New York, St. Louis, San Francisco, Auckland, Bogota, Caracas, Lisbon, London, Madrid, Mexico City, Milan, Montreal, New Delhi, San Juan, Singapore, Sudney, Tokyo, Toronto, 1291.
- Microdeletions and Molecular Genetics, Annick VOGELS and Jean-Pierre FRYNS. Center for Human, University of Leuven, Herestraat 49, B-3000 Leuven, Belgium, 2004-02.
- Di George, A. 1965, A new concept of the cellular basis of immunity. J. Pediatr., 67, 907.
- Clinical spectrum and molecular diagnosis of Angelman and Prader-Willi syndrome patients with an imprinting mutation.Saitoh S, Buiting K, Cassidy SB, Conroy JM, Driscoll DJ, Gabriel JM, Gillessen-Kaesbach G, Glenn CC, Greenswag LR, Horsthemke B, Kondo I, Kuwajima K, Niikawa N, Rogan PK, Schwartz S, Seip J, Williams CA, Nicholls RD, Am J Med Genet. 1997;68(2):195.
Источник