Код по мкб синдром сильвера рассела

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Причины
- Симптомы
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Синдром Рассела-Сильвера.
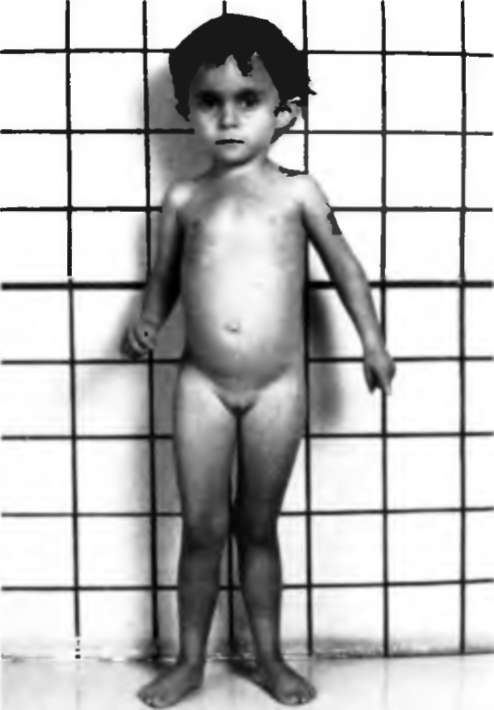
Синдром Рассела-Сильвера
Описание
Синдром Рассела-Сильвера врожденное заболевание, характеризующееся внутриутробной задержкой роста плода, поздним закрытием большого родничка, нарушением формирования скелета. Тип наследования неизвестен. Встречается в большинстве случаев спорадически, однако описаны и единичные родословные с данной патологией. Частота в популяции 1:30 000. Встречается в равной степени, как у мужчин, так и у женщин.
Причины
Причиной заболевания считаются генетические нарушения, приводящие к подобным порокам развития. Дети с данной патологией имеют малую длину и массу тела при рождении. Значительная задержка роста и костного созревания наблюдается и в дальнейшем.
Главной причиной преждевременного полового созревания при этом заболевании является избыток гонадотропных гормонов.
Симптомы
Дети рождаются небольшой длины (до 45 см) и с малой массой тела (1,5—2,5 кг). С годами отставание в росте сохраняется, в связи, с чем окончательный рост у женщин менее 150 см, у мужчин немногим выше 150 Масса тела у взрослых нормальная или даже избыточная. Часты аномалии мочевыделительной системы и наружных половых органов: крипторхизм, гипоспадия, гипоплазия полового члена, мошонки. Характерна асимметрия тела (лицо, туловище, длина ног). Лицо треугольной формы: псевдогидроцефалия, большой лоб и гипоплазия нижней челюсти, высокое нёбо, нередко с расщелиной, оттопыренные уши. Узкая грудная клетка, короткие руки, поясничный лордоз. Половое развитие опережает возраст ( в 30% случаев), начинается в 5-6 лет и обусловлено гонадотропной стимуляцией яичников. Интеллект сохранен в большинстве случаев.
Лечение
С 1985 г. Для лечения детей с соматотропной недостаточностью используются исключительно генно-инженерные препараты гормона роста человека.
В настоящее время в России прошли клиническую апробацию и разрешены к использованию следующие рекомбинантные препараты гормона роста человека: Нордитропин RНордиЛетR (Ново Нордиск, Дания); хуматроп (Лилли Франс, Франция); генотропин (Пфайзер Хелс АБ, Швеция); сайзен (Индустрия Фармасьютика Серано С. А. , Италия); растан (Фармстандарт, Россия).
При лечении гипофизарного нанизма у детей имеется четкая связь «доза–ростовой эффект», особенно выраженная в первый год лечения. Рекомендуемая стандартная доза СТГ при терапии классического дефицита СТГ – 0, 033 мг/кг/на инъекцию, ежедневно, подкожно, в вечернее время 20,00-22,00.
Критерием эффективности терапии является увеличение скорости роста от исходной в несколько раз. Она достигает в первый год лечения, по данным разных авторов, от 8 до 13 см в год. Максимальная скорость роста отмечается в первый год лечения, особенно в первые 3-6 мес, затем имеет место замедление скорости роста от первого ко второму году лечения (при сохранении скорости роста более 5-6 см в год).
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Причины
- Патогенез
- Симптомы
- Лечение
Другие названия и синонимы
Синдром гипотонии-ожирения.
Названия
Синдром Прадера — Вилли.
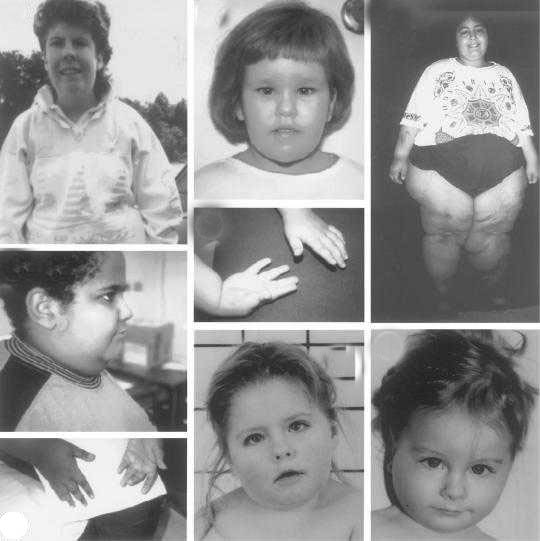
Проявления синдрома Прадера — Вилли
Синонимы диагноза
Синдром гипотонии-ожирения.
Описание
Синдром Прадера — Вилли — редкая генетическая аномалия. При синдроме Прадера — Вилли отсутствуют или не экспрессируются примерно 7 генов из 15-й хромосомы, унаследованной от отца.
Кариотип 46 XX или ХУ, 15q-11-13. Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г.
По данным регистра ассоциации больных с синдромом Прадера-Вилли, в США и Канаде на декабрь 1986 г. Насчитывалось 1595 больных. В последние годы удалось установить популяционную частоту патологии, составляющую 1 : 10 000 — 1 : 20 000.
Причины
Авторы, впервые описавшие синдром, высказывали предположение об аутосомно-рецессивном типе наследования заболевания. Затем появились сообщения о возможности аутосомно-доминантной передачи болезни. Подтверждением данных гипотез могли служить наблюдавшиеся семейные случаи патологии. Однако большинство описанных клинических наблюдений синдрома Прадера — Вилли носило спорадический характер.
Последующие исследования позволили установить у детей с синдромом Прадера — Вилли определенные хромосомные нарушения. Цитогенетический анализ показал, что хромосомные аномалии у больных были представлены либо транслокациями (t 15/15), либо мозаицизмом. В 1987 г. Появились первые сообщения о микроделеции хромосомы 15. Однако окончательная идентификация хромосомных изменений при синдроме Прадера — Вилли стала возможной только после внедрения в практику молекулярно-генетических методов исследования.
В настоящее время установлено, что развитие синдрома Прадера — Вилли связано с повреждением критического района хромосомы 15 (сегмента q11,2- q13). При этом оказалось, что повреждение этого же участка хромосомы 15 наблюдается и при другом заболевании — синдроме Ангельмана, клиническая картина которого существенно отличается от синдрома Прадера — Вилли и характеризуется ранним (в возрасте 6-12 мес) замедлением психомоторного развития, микроцефалией, нарушением речи (в 100% случаев), атаксией, неконтролируемым насильственным смехом, частыми эпилептиформными припадками, специфическим выражением лица.
Таким образом, несмотря на повреждение при синдромах Прадера — Вилли и Ангельмана одного и того же локуса хромосомы 15, клинические проявления обеих болезней резко противоположны.
Объяснение фенотипических различий получено лишь в последние годы. Оказалось, что развитие этих заболеваний связано с новыми генетическими явлениями — геномным импринтингом и унипарентальной дисомией.
Геномный импринтинг — новое явление, открытое благодаря успехам молекулярной генетики. Он означает различную экспрессию генетического материала (гомологичных аллелей) в хромосомах в зависимости от отцовского или материнского происхождения, т. Е. Свидетельствует о влиянии родителей на фенотип ребенка. До настоящего времени считалось, что вклад в проявляемость (экспрессию) генов отца и матери равноценен.
По сути геномный импринтинг — это половой и тканевозависимый сложный модификатор генной активности некоторых локусов хромосом в зависимости от их родительского происхождения. Проявления геномного импринтинга выявлены и при других заболеваниях — синдромах Сотоса, Беквита-Видемана, Сильвера-Рассела, муковисцидозе и других.
Унипарентальная (однородительская) дисомия — наследование обеих хромосом только от одного из родителей. В течение многих лет считалось, что такое наследование невозможно. Лишь с помощью молекулярно-генетических маркеров удалось доказать возможность однородительской дисомии. Природа унипарентальной дисомии окончательно не выяснена, однако установлено, что она обязана своим происхождением ряду генетических и биохимических нарушений.
Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить микроделецию или унипарентальную дисомию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы — прометафазный анализ, использование ДНК-маркеров определенных участков хромосомы 15 (исследование процессов метилирования) и.
На сегодняшний день синдромы Прадера — Вилли и Ангельмана служат общепринятой моделью для изучения новых в клинической генетике и сложных явлений — геномного импринтинга и унипарентальной дисомии.
Установлено, что синдром Прадера — Вилли может быть обусловлен двумя основными механизмами. Первый из них — микроделеция хромосомы 15 (15q11,2-q13), которая всегда отцовского происхождения. Второй — материнская изодисомия, т. Е. Когда обе хромосомы 15 получены от матери. Развитие синдрома Ангельмана, наоборот, связано с микроделецией того же участка хромосомы 15, но материнского происхождения, или отцовской изодисомией. Большинство (около 70%) случаев синдрома Прадера — Вилли обусловлено микроделецией, остальные — дисомией. При этом обращает на себя внимание отсутствие клинических различий между больными с микроделецией и изодисомией.
Патогенез
Патогенез синдрома Прадера — Вилли до настоящего времени остается малоисследованным. Высказываются предположения, что ожирение у больных обусловлено значительным (более чем в 10 раз) усилением синтеза жира из ацетата и крайне низкими процессами липолиза.
Гипогонадизм по гипогонадотропному типу может быть связан с дисфункцией гипоталамуса, преимущественно, в области вентромедиального и вентролатерального ядер. Правильность данной точки зрения подтверждается эффективностью лечения больных фармацевтическими препаратами (кломифен), приводившими к увеличению в плазме содержания лютеинизирующего гормона, тестостерона, нормализации показателей почечной экскреции гонадотропинов, сперматогенеза и появлению вторичных половых признаков.
Одним из объяснений гипопигментации кожи, волос и радужки служит снижение активности тирозиназы в волосяных фолликулах и меланоцитах, а также уменьшение пигмента в сетчатке.
Обращается внимание на повышенный риск развития лейкемии у больных с синдромом Прадера — Вилли. Исследования выявили снижение репарации ДНК (до 65% по сравнению с 97% у здорового ребенка) в лимфоцитах больных с данной патологией. Не исключено, что низкая репарационная способность ДНК может играть роковую роль в развитии злокачественных новообразований у лиц с синдромом Прадера — Вилли.
Симптомы
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание.
В течение заболевания можно выделить две фазы: первая — свойственна детям 12-18 мес жизни. Она характеризуется выраженной мышечной гипотонией, снижением рефлексов — Моро, сосательного и глотательного, что затрудняет кормление ребенка. Вторая — наступает позже, через несколько недель или месяцев. Появляются полифагия, постоянное чувство голода, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей.
Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает. Стопы и кисти больных диспропорционально маленькие — акромикрия. У детей отмечается гипогонадизм (у мальчиков — гипоплазия полового члена, мошонки, крипторхизм, а у девочек — недоразвитие половых губ и в 50% случаев — матки).
Рост больных нередко снижен. У 75% детей наблюдается гипопигментация кожи, волос и радужки. Часто диагностируется микроцефалия. Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед. ). Речь затруднена, словарный запас уменьшен. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, страбизм.
Встречаются и другие аномалии: микродонтия, гипоплазия хрящей ушных раковин, сколиоз, эктропион (выворот века), глаукома.
Нередко развитие сахарного диабета, который с возрастом имеет тенденцию к улучшению.
При морфологическом исследовании мозга и ЯМР-томографии могут наблюдаться (примерно в 12% случаев) кисты червя мозжечка, аномалии коры головного мозга.
Продолжительность жизни больных может достигать 60 лет и более.
Лечение
Терапия синдрома Прадера — Вилли окончательно не разработана. По данным литературы, комплекс лечебных мероприятий включает лишь диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины).
Источник
Современной медицине известно достаточно много заболеваний. Некоторые из них изучены довольно хорошо, над причинами и методами лечения других безуспешно работают группы ученых. Часть заболеваний приобретенные, другие же являются врожденными. Одним из таких врожденных заболеваний и является синдром Рассела-Сильвера.
Особенности заболевания
Существуют и другие популярные названия: карликовость Сильвера-Рассела, ССР.
Рассел А. и Х. К. Сильвер — врачи-педиатры, которые занимались изучением пренатальной задержки в развитии.
Синдром Рассела-Сильвера является заболеванием врожденным. Главной его особенностью является задержка физического развития еще во время беременности, в частности, нарушено формирование скелета ребенка. В дальнейшем может наблюдаться позднее закрытие родничка.
Причины наследования до сих пор неизвестны, в большинстве случаев нет определенной системы.

Данное заболевание встречается у одного человека на 30 000. Половая принадлежность на развитие болезни не влияет.
Причины возникновения синдрома Рассела-Сильвера
Главной причиной заболевания являются исключительно нарушения на уровне генетики. Причем форма наследования не носит периодический или системный характер.
Чаще всего страдают хромосомы 7 (10% случаев), 11, 15, 17, 18. Именно эти хромосомы и отвечают за рост человека. В большинстве случаев это происходит из-за того, что ребенок наследует две копии хромосомы от матери. Данный эффект носит название однородительской материнской дисомии.
Внешние симптомы заболевания
При рождении ребенок с синдромом Рассела-Сильвера имеет довольно маленькую массу, обычно не более 2500 г, хотя беременность и считается доношенной. Длина при этом около 45 см. С возрастом эта проблема не решается и отставание в росте наблюдается и у взрослых людей (у женщин рост не более 150 см, у мужчин чуть более 150 см). Однако масса полностью соответствует возрасту, в некоторых случаях даже превышает норму.

Претерпевает изменения и мочеполовая система, например, наблюдается крипторхизм (нарушение, при котором яички расположены не на своих местах), гипоспадия (мочеиспускательный канал открывается в нетипичном для этого месте), гипоплазия полового члена и мошонки (недоразвитость).
Внешне синдром Рассела-Сильвера также наблюдается. Выражается он в ассиметрии тела. Затрагивает это и лицо, и туловище, и длину ног и рук.
Влияет синдром Рассела-Сильвера (лечение заболевания можно узнать из статьи) и на лицо. Часть черепной коробки, в которой располагается головной мозг, увеличена по сравнению с лицевой ее частью, причем увеличение явно непропорционально. Форма лица напоминает треугольник, при котором лоб выпуклый, а размеры нижней челюсти и рта значительно уменьшены. Это называется псевдогидроцефалия. Губы узкие, а уголки слегка опущены (эффект «рот карпа»). Небо высокое, в некоторых случаях может быть с расщелиной. Уши в большинстве случае оттопырены.
Среди сопутствующих внешних симптомов можно выделить:
- нарушение формирования подкожного жира;
- узкая грудная клетка;
- лордоз в области поясницы (выпуклость позвоночного столба вперед);
- искривления мизинца.
Сопутствующие заболевания внутренних органов
Помимо внешних расстройств, часто наблюдаются и внутренние проблемы организма. Синдром Рассела-Сильвера (симптомы, связанные с нарушениями внешнего вида, указаны ранее) влияет на работу почек в связи с их неправильной сформированностью (подковообразная форма, расширение почечной лоханки, ацидоз канальцев).
Для больных независимо от их половой принадлежности характерно раннее половое созревание. В 30% случаев начинается в возрасте около 6 лет. Это напрямую связано с тем, что происходит гонадотропная стимуляция яичников (количество половых гормонов значительно увеличено).
А вот интеллект полностью сохранен.
Синдром Рассела-Сильвера: диагностика
Диагностируется данное заболевание уже в раннем детстве. Такое решение принимает педиатр, наблюдающий больного ребенка. Однако помимо обычного наблюдения проводятся и различные лабораторные анализы и тесты:
- Определение уровня сахара в крови. Очень часто дети, которым ставится диагноз «синдром Рассела-Сильвера», имеют пониженный уровень глюкозы в крови.
- Тестирование на хромосомные аномалии. В большинстве случаев эти проблемы обнаруживаются.
- Определение количества гормона роста. При данном заболевании наблюдается его дефицит.
- Обследование сформированности скелета. Это требуется для того, чтобы исключить полностью дополнительные условия, которые в некоторых случаях могут давать ложный положительный результат.

Особенности лечения
Главное правило лечения: своевременная диагностика. Если не сделать этого вовремя, врач может пойти по ложному пути и заниматься лечением гидроцефалии, однако у таких детей этого заболевания нет.
В большинстве случаев таким больным назначается прием гормона роста по определенной схеме, которую разрабатывает лечащий врач.
Помимо этого часто используются и дополнительные методы:
- физиотерапия, которая направлена на улучшение состояния мышц;
- специальное образование.
В процессе лечения принимают одновременно несколько специалистов:
- врач-генетик, который способен выявить данное заболевание в самом его начале;
- диетолог или гастроэнтеролог, главное задание которого разработать особую диету, что направлена на повышение роста;
- эндокринолог, который и назначает гормон роста;
- психолог.
Определить эффективность проведенного лечения можно по увеличению скорости роста. При правильно разработанной схеме уже в первый год лечения можно достичь результата в 8 см.
Источник
В современной медицинской практике существует большое количество различных заболеваний. Одни являются хорошо известными, приобретаемыми в процессе жизнедеятельности, другие — врожденными. Синдром Рассела-Сильвера — это одна из генетических болезней.

Особенности заболевания
Известно и другое название данного синдрома, такое как карликовость Сильвера-Рассела. Исследованием внутриутробной задержки в развитии занимались врачи-педиатры А. Рассел и Х. К. Сильвер, в честь которых и названо заболевание.
Характерной чертой этого недуга можно назвать задержку физического развития еще в период вынашивания плода, а именно нарушение формирования скелета ребенка. Впоследствии наблюдается позднее закрытие родничка. Наследственные причины пока еще недостаточно исследованы. Заболевает данным синдромом один человек на 30 тысяч. Гендерные признаки на течение заболевания не влияют. Патогенез синдрома Рассела-Сильвера рассмотрим ниже.
Причины возникновения
Первопричиной заболевания являются изменения на генетическом уровне, при котором характер наследования не является периодическим или системным. В основном подвергаются нарушениям хромосомы семь (десять процентов случаев), одиннадцать, пятнадцать, семнадцать, восемнадцать, которые являются основополагающими при формировании роста человека. Маленький рост чаще всего возникает из-за того, что ребенок наследует эти хромосомы от матери. Этот процесс называется однородительская материнская дисомия. Ребёнок, который рождается с этой болезнью, имеет очень маленький вес тела (не более 2500 г), несмотря на то, что беременность является доношенной. В процессе жизнедеятельности задержка роста отмечается и у взрослых людей. Вес тела полностью соответствует возрасту, иногда бывает больше нормы.
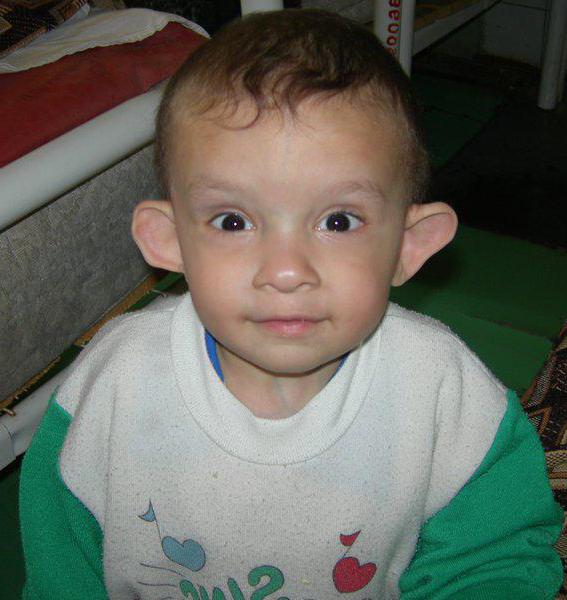
Симптомы синдрома Рассела-Сильвера
Нарушается работа и мочеполовой системы, связанная с крипторхизмом (изменение в половой системе, когда яички находятся в нетипичном для этого месте), гипоспадия (мочеиспускательный канал расположен не на своём месте), недоразвитость полового члена и мошонки. Внешние изменения человека с этим заболеванием имеют место, что проявляется в асимметрии тела, это касается и лица, и тела, длины рук и ног.
Оказывает влияние синдром Рассела-Сильвера и на форму лица. Мозговой отдел черепной коробки, где находится головной мозг, по размерам намного больше, чем её лицевая часть, при этом наблюдается явное увеличение с нарушением пропорций. Лицо принимает треугольный вид, где лоб выпуклый, а нижняя челюсть и рот в значительной степени меньше, такое изменение носит название псевдогидроцефалия. Уголки губ немного опущены вниз, нёбо приподнято, иногда оно встречается даже с расщелиной. Уши у больного синдромом чаще всего оттопырены. Кроме, этого к внешним признакам можно отнести: нарушение выделения сальных желёз, узкая грудная клетка, выпуклая форма позвоночного столба вперед; искривление мизинца.
Заболевания внутренних органов, которые сопутствуют синдрому
Кроме внешних признаков, таких как маленький рост и искажение черт лица, также необходимо отметить и проблемы с внутренними органами. Данный синдром (признаки, указывающие на нарушения внешнего вида, представлены ранее) оказывает влияние на функционирование почек в связи с тем, что они были неправильно сформированы (форма подковы с расширенной почечной лоханкой и ацидозом канальцев). Наблюдаются изменения и со стороны половой системы: независимо от пола свойственно половое созревание на ранних этапах. У тридцати процентов людей в возрасте около шести лет. Связано это с тем, что происходит значительное увеличение количества половых гормонов, однако интеллектуальные способности находятся в норме.
Диагностика синдрома Рассела-Сильвера
Данное заболевание диагностируется еще в детском возрасте. Такой диагноз ставит педиатр при осмотре больного ребенка. Кроме стандартного осмотра пациент сдаёт анализы для лаборатории для определения уровня содержания сахара в крови. Являются частыми случаи, когда у пациентов, которым ставится такой диагноз, наблюдается пониженный уровень глюкозы в крови. Также проводится тестирование на определение хромосомных аномалий. Чаще всего эти проблемы имеют место быть.
Больному необходимо сдать тест на количество гормонов роста, так как при данном синдроме наблюдается его недостаток. Необходимо также провести анализ сформированности скелета, для того, чтобы исключить факторы, которые могут дать ложный положительный результат. Диагноз в основном является клиническим, но он может подтверждаться генетической аномалией.
Особенности лечения
Так же как и основное количество наследственных заболеваний, синдром Рассела-Сильвера не подразумевает особенного лечения. Все методы терапии при данном заболевании сконцентрированы на обеспечении лучших жизненных условий. При замедленном росте больным прописывается гормон роста. Относительно раннего полового созревания назначаются специальные гормональные препараты.
Встречаются отдельные случаи, когда при данном недуге необходимо прибегнуть к помощи пластического хирурга. Кроме этого, иногда больного ребенка приходится переводить на индивидуальное обучение в связи с тем, что у ребенка наблюдается умственная отсталость, однако интеллект при данном синдроме в норме. Применять данные меры приходится из-за психологических расстройств у ребенка в процессе его обучения в обычной школе.
С диагнозом синдром Рассела-Сильвера больные должны быть на учёте у врача-эндокринолога, регулярно проходить медосмотры.

Женщины могут как зачать ребёнка, так и выносить его. Вероятность наследственности данного заболевания является индивидуальным в каждом отдельном случае.
Если девушка не планирует беременность, и ее беспокойства ограничиваются только избыточным оволосением, гнойными высыпаниями на коже и нарушением менструального цикла, то необходимо пройти терапию лекарствами, содержащими антиандрогены и эстрогены.
Однако использование глюкокортикоидных препаратов, восстанавливающих работу яичников, практически не оказывает влияния на уменьшение избытка волос. В случае явного вирилизма половых органов проводят пластическую корректирующую операцию, которая заключается в удалении адреногенитальных признаков, которые могут проявляться от гипертрофии клитора до полной маскулинизации гениталий.

Кроме этого, если причиной вирилизма является опухоль в надпочечнике или яичнике, то рекомендован хирургический метод лечения. Сколько живут с синдромом Сильвера-Рассела? Рассмотрим далее.
Прогноз
У больного человека наблюдаются небольшой рост и вес, в остальном долгосрочный прогноз будет хорошим.
Источник