Код мкб синдром картагенера

Синдром Картагенера – наследственное заболевание, которое является формой цилиарной дискинезии и включает в себя три компонента: обратное расположение легких, хронические бронхоэктазы и патологии пазух носа.
Причины
Синдром Картагенера встречается у 1 из 30-50 тысяч новорожденных. Впервые триаду симптомов описал швейцарский врач Зиверт Картагенер в 1933 году. Его исследования указывали на наследственную природу заболевания.
Синдром Картагенера у детей возникает из-за мутаций в генах, которые отвечают за формирование реснитчатого эпителия, выстилающего слизистые оболочки дыхательных путей. Как правило, заболевание провоцируют дефекты в генах DNAI1 и DNAH5. Передается патология по аутосомно-рецессивному принципу.
Генетические изменения приводят к нарушению функций реснитчатого эпителия: ворсинки либо вообще не двигаются, либо работают асинхронно. В результате существенно нарушается функционирование дыхательной системы: из-за сбоя в механизме самоочищения возникают хронические воспалительные процессы – бронхиты, синуситы, отиты, евстахииты и так далее.
Симптомы
Основное проявление синдрома Зиверта Картагенера – подверженность ребенка респираторным заболеваниями: ринитам, синусита, бронхитам, пневмониям. Рецидивирующее воспаление дыхательных путей со временем приводит к деструкции нервно-мышечного слоя бронхов и их сегментарному расширению (бронхоэктазам). Кроме того, синдром Картагенера характеризуется такими признаками, как:
- отставание в физическом развитии;
- вялость, периодические головные боли, приступы тошноты, потливость;
- повышение температуры в период обострений;
- кашель с отхождением гнойной мокроты;
- ухудшение носового дыхания;
- выделения из носа с примесями гноя;
- потеря обоняния;
- полипы в носовых ходах;
- хронический отит, приводящий к снижению слуха;
- утолщение фаланг пальцев и деформация ногтей из-за ухудшения кровообращения в конечностях (после 6-7 лет);
- бледность кожи и посинение носогубного треугольника при нагрузке.
Один из компонентов синдрома Картагенера – обратное расположение легких. У 50% детей оно сочетается с правосторонней локализацией сердца и зеркальным расположением других внутренних органов.
Существует взаимосвязь между активностью ресничек эпителия и подвижностью жгутиков сперматозоидов, поэтому при синдроме Картагенера наблюдается мужское бесплодие. Также данное заболевание зачастую сочетается с гипогинезией лобных пазух, полидактилией, аномалиями строения мочевыводящих путей, гипофункцией эндокринных органов, поражением сетчатки и другими патологиями.
Диагностика
Диагностика синдрома Картагенера включает такие направления, как:
- сбор анамнеза;
- физикальный осмотр, перкуссия и аускультация – обнаруживаются обратное расположение органов, жесткое дыхание, хрипы;
- рентгенография – показывает усиление рисунка по сторонам бронхов и затемнение придаточных пазух носа;
- бронхоскопия – демонстрирует наличие гнойной мокроты;
- бронхография – визуализирует бронхоэктазы;
- исследования крови (общий анализ, биохимия, иммунограмма).
Обязательно проводится осмотр отоларингологом, который подтверждает наличие хронического синусита и отита.
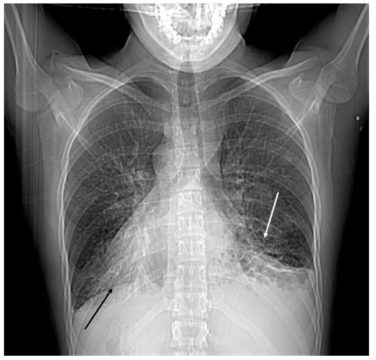 Рентгенография легких при синдроме Картагенера.
Рентгенография легких при синдроме Картагенера.
Лечение
Поскольку синдром Картагенера является генетическим заболеванием, его этиотропное лечение не разработано. Терапия направлена на облегчение симптомов.
Основные методы лечения:
- физиопроцедуры – дренажный массаж, ЛФК;
- противовоспалительная терапия и улучшение функционирования бронхов – ингаляции или бронхоскопия с муколитиками и бронхоспазмолитиками;
- введение антибиотиков в период обострений, которые назначаются с учетом выявленной чувствительности инфекционных агентов;
- прием иммуномодуляторов и витаминов;
- введение плазмы и иммуноглобулинов;
- санация придаточных пазух носа;
- удаление расширенных участков бронхов.
Прогноз
Синдром Картагенера имеет относительно положительный прогноз при условии адекватного лечения. На неблагоприятное течение заболевания указывают развитие выраженной дыхательной недостаточности и значительная интоксикация организма ребенка.
Профилактика
Синдром Картагенера возникает в результате генетических мутаций, поэтому предупредить его развитие невозможно.
Источник
Первичная цилиарная дискинезия — это генетически детерменированное заболевание, в основе которого лежит дефект ультраструктур ресничек мерцательного эпителия, приводящий к нарушению их двигательной функции[1].
Этиология и распространенность[править | править код]
Тип наследования — аутосомно-рецессивный, описаны и более редкие Х- сцепленные формы.
ПЦД относится к орфанным заболеваниям[2], по данным разных авторов распространенность составляет от 1:15000 до 1:60000 новорожденных. Наиболее частой и классической формой является синдром Картагенера (40 — 60% от общего числа)[3].
Симптомы[править | править код]
Симптом барабанных палочек
Ведущим проявлением первичной цилиарной дискинезии являются рецидивирующие с детского возраста воспалительные заболевания верхних и нижних дыхательных путей[4], постоянный кашель с гнойной мокротой, влажные хрипы в легких[5]. В более позднем возрасте формируются бронхоэктазы. Можно наблюдать отставание в физическом развитии, изменение концевых фаланг пальцев (симптом барабанных палочек). У многих больных отмечаются рецидивирующие экссудативные отиты со снижением слуха[4].
При синдроме Картагенера помимо бронхоэктазов и синусита имеется обратное расположение внутренних органов или изолированная декстрокардия[3].
Другие проявления[править | править код]
У мужчин с ПЦД часто выявляют бесплодие, вызванное дефектной подвижностью сперматозоидов, у женщин нередко наблюдается внематочная беременность, вызванная аномальным строением фимбрий фаллопиевых труб.
Крайне редко аномалия ресничек приводит к пигментной ретинопатии, биллирному циррозу печени, внутрненней гидроцефалии и поликистозу почек[4].
Диагностика[править | править код]
- исследование уровня оксида азота в выдыхаемом назальном воздухе;
- анализ частоты и паттерна биения ресничек в биоптате из полости носа или бронха с помощью световой микроскопии;
- обнаружение аномалий строения ресничек в биоптате слизистой оболочки носа или бронха с помощью электронной микроскопии[4].
Лечение[править | править код]
Терапия в первую очередь направлена на предупреждение прогрессирования бронхоэктазов и сохранение нормальной легочной функции, а также носового дыхания и слуха[4].
Консервативное лечение[править | править код]
- курсовое лечения муколитическими препаратами;
- при наличии бронхообструктивного синдрома — ингаляционные бронхолитики;
- при обострении хронического бронхолегочного процесса рекомендуется назначение антибиотиков;
- промывание носовых ходов гипертоническим раствором натрия хлорида, применение назального душа[4].
Хирургическое лечение[править | править код]
- оперативное лечение нижних дыхательных путей рекомендуется крайне редко (описаны случаи пациентов, которым проводилась трансплантация легких[6]);
- при частом обострении хронического синусита рекомендуется рассмотреть вопрос о проведении операции с целью улучшения аэрации и дренажа параназальных синусов.
Примечания[править | править код]
- ↑ Ж. А. Безлер, В. И. Бобровничий. Первичная цилиарная дискинезия (other). — 2011. (недоступная ссылка)
- ↑ С. Ю. Елизарова, О. В. Сидорович, А. В. Хижняк, И. В. Королева, И. В. Рыбакова. КЛИНИЧЕСКИЙ СЛУЧАЙ РАННЕЙ ДИАГНОСТИКИ И ЛЕЧЕНИЯ ПЕРВИЧНОЙ ЦИЛИАРНОЙ ДИСКИНЕЗИИ (СИНДРОМА КАРТАГЕНЕРА). Архивъ внутренней медицины (12 августа 2018). Дата обращения 22 марта 2019.
- ↑ 1 2 Шинкарёва В.м, Павлова Т.б. Первичная цилиарная дискинезия. Клиническое наблюдение // Acta Biomedica Scientifica. — 2016. — Т. 1, вып. 1 (107). — ISSN 2541-9420.
- ↑ 1 2 3 4 5 6 Первичная цилиарная дискинезия у детей. Клинические рекомендации. Министерство здравоохранения Российской Федерации (2016).
- ↑ 7. Первичная цилиарная дискинезия (синдром неподвижных ресничек) и синдром Картагенера. Госпитальная педиатрия: конспект лекций.
- ↑ Orphan lung diseases. — Plymouth: European Respiratory Society, 2011. — С. 201—217. — viii, 365 pages с. — ISBN 9781849840132, 184984013X.
Источник
Синдром Картагенера (Kartagener syndrome, синдром неподвижных ресничек, двигательная цилиопатия, триада Картагенера) — разновидность генетически обусловленного аутосомно-рецессивного расстройства — первичной цилиарной (реснитчатой) дискинезии, характеризующейся цилиарной дисфункцией и нарушением мукоцилиарного клиренса.
673 •
•
21.02.2019
Иллюстрация Георгия Сапего
Впервые детальное описание синдрома было дано в 1933 году М. Картагенером. Именно он распознал эту клиническую триаду как целостный синдром. Хотя до него еще в 1904 году А.К. Зиверт описал бронхоэктазы в сочетании с декстрокардией.
В последние годы выявлено значительное количество генов и их мутаций, которые могут лежать в основе заболевания. Тип наследования — чаще аутосомно-рецессивный, но на сегодняшний день описаны и более редкие Х-сцепленные формы заболевания.
Лишь в 1976 г. было обнаружено, что в основе заболевания лежит неподвижность ресничек эпителия из-за дефекта их ультраструктуры, называемая первичной дискинезией цилиарного эпителия (ДЦЭ). Реснички представляют собой крошечные, похожие на волосы структуры, которые обнаруживаются на поверхности клеток в различных частях тела.
Как правило, синдром обусловлен отсутствием или дефектами строения внутренних и наружных динеиновых ручек в структуре ресничек и жгутиков. Могут обнаруживаться дефекты радиальных спиц и микротрубочек, а также возможны случаи полного отсутствия ресничек. У некоторых больных имеются сочетания нескольких дефектов. Также реснички и жгутики могут иметь и нормальную ультраструктуру, однако при этом, как правило, определяется аномалия белка тяжелой цепи аксонемального динеина.
Координированные движения ресничек необходимы для нормального функционирования многих органов и тканей. Кроме того, движение ресничек помогает установить право-левую ось во время эмбрионального развития.
К основным симптомам заболевания относятся: бронхоэктазы, хронический кашель, носовые полипы, синусит, аносмия, дефекты роговицы, средний отит, постоянные головные боли, транспозиция внутренних органов, гипогаммаглобулинемия А, пониженная подвижность лейкоцитов. Нарушения движения ресничек приводят и к другим клиническим проявлениям. В частности, описаны случаи гидроцефалии у пациентов с синдромом Картагенера..
В настоящее время нет единого метода диагностики синдрома Картагенера. При установлении диагноза учитываются: клиническая картина; исследование уровня оксида азота (NO) в выдыхаемом назальном воздухе; анализ частоты и паттерна биения ресничек в биоптате из полости носа или бронха с помощью световой микроскопии; электронная микроскопия. Для подтверждения диагноза рекомендовано исследование паттерна и частоты биения ресничек в сочетании с электронной микроскопией у пациентов с поражением верхних и нижних дыхательных путей в состоянии ремиссии длительностью не менее месяца.
Специфической терапии синдрома Картагенера не разработано. Лечение варьирует в зависимости от признаков и симптомов, присутствующих у каждого человека.
К современным методам лечения можно отнести следующий ряд процедур:
Применение антибиотиков
Использование постурального массажа
Физиотерапия
Прием иммуномодуляторов и витаминов
Прогноз при раннем выявлении заболевания и своевременно начатом лечении благоприятен. Сегодня также ученые занимаются вопросом о репродуктивной функции больных с синдромом Картагенера. Известно, что структурные изменения в жгутиках сперматозоидов и ресничках маточных труб аналогичны таковым в ресничках дыхательных путей. Это приводит к мужскому бесплодию и не исключает риск бесплодия у женщин при данной патологии.
Частота встречаемости от 1 на 2265 до 1 на 40000 населения.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Ретта.
Синдром Ретта
Описание
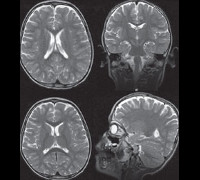
Синдром Ретта. Генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника. Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного.
Дополнительные факты
Синдром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году. Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера.
Синдром Ретта
Причины
Этиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается. Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани.
При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей. Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК. В результате этого у больных синдромом Ретта наблюдается значительное превалирование процессов торможения в головном мозге, что отражается не только на физиологии центральной нервной системы, но и на ее морфологическом строении.
Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко. Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца. Дефекты этого гена приводят и другим патологиям центральной нервной системы – варианту Запела, синдрому Луба (Х-сцепленная умственная отсталость у мальчиков), врожденной энцефалопатии. Некоторые исследователи относят эти состояния к атипичным формам синдрома Ретта.
Симптомы
У новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям. Врачи-педиатры отмечают более медленный, нежели в норме, рост стоп и кистей в длину и замедление роста окружности головы. Иногда помимо неврологических проявлений может отмечаться нарушение работы печени, сердца, желудочно-кишечного тракта.
Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе. Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения.
Судороги. Эхолалия.
Диагностика
Диагностика синдрома Ретта производится на основании изучения анамнеза больного, его настоящего статуса, магнитно-резонансной томографии и энцефалографии, молекулярно-генетических анализов. Изучение наследственного анамнеза, как правило, не имеет особого смысла по причине спорадического характера мутаций гена MECP2. Характерными для синдрома Ретта являются нормальное развитие ребенка до 6-12 месяцев, возникновение мышечной гипотонии и беспокойства в раннем детстве, появление в дальнейшем атаксии и частых эпилептических припадков, стремительное утрачивание приобретенных навыков. В дальнейшем у больных регистрируется тяжелая умственная отсталость, мышечная слабость (вплоть до атрофии), искривление позвоночника, судорожные припадки.
При осмотре больных синдромом Ретта выявляется отставание в росте и его остановка, резкое уменьшение окружности головы, отсутствие речи (на начальных этапах патологии характерна эхолалия). На магнитно-резонансной томографии головного мозга обнаруживается уменьшение размера органа, нечеткая дифференциация серого и белого вещества, базальных ганглиев, снижение складчатости коры больших полушарий. Электроэнцефалограмма подтверждает снижение фоновой активности головного мозга и резко ослабленную реакцию на внешние раздражители. Наиболее точную диагностическую информацию дают методы современной генетики – поиск делеций в локусе гена MECP2 или прямое секвенирование его последовательности для определения мутаций. Такое подтверждение синдрома Ретта возможно и в рамках пренатальной диагностики генетических заболеваний. Вспомогательную роль в установлении этого состояния может играть обследование внутренних органов (например, методами УЗИ) – у 20-30% больных выявляется недоразвитие печени или селезенки.
Лечение
Специфического лечения синдрома Ретта на сегодняшний день не существует. Имеются обнадеживающие данные некоторых исследовательских лабораторий, сотрудникам которых удалось «включить» ген MECP2 у мышей и тем самым добиться исчезновения симптомов заболевания. В сфере практической медицины пока доступна только симптоматическая терапия, однако и она сопряжена с рядом трудностей – в частности, эпилептические припадки при этом заболевании плохо поддаются устранению противосудорожными средствами. Также для лечения синдрома Ретта применяют ноотропные препараты, нарушения сна корректируют снотворными препаратами из группы барбитуратов или мелатонином.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник