Генетические синдромы с поликистозом почек

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 26 октября 2017;
проверки требуют 7 правок.
Поликисто́з почек (синоним: поликистозная болезнь почек, сокр. ПБП) представляет собой генетическое заболевание, проявляющееся кистозным перерождением паренхимы почек. Одна из форм поликистозной дисплазии почек.[2] Болезнь затрагивает не только сами почки, но часто также другие органы (печень).
Классификация[править | править код]
У человека выделяют две основные формы поликистозной болезни почек, различающиеся по типу наследования: аутосомно-рецессивная поликистозная болезнь почек (характерна для детского возраста) и аутосомно-доминантная поликистозная болезнь почек (чаще начинает проявляться в возрасте 30-50 лет).[3]
Аутосомно-рецессивная поликистозная болезнь почек связана с мутацией гена PKHD1, который кодирует белок фиброцистин.[4]
Аутосомно-доминантная поликистозная болезнь почек встречается в человеческой популяции с частотой 1/400 — 1/1000, являясь одной из наиболее распространённых генетических болезней. Общее количество больных в мире оценивается в 10-12 млн человек. 85 % случаев аутосомно-доминантного поликистоза почек вызваны мутацией гена PKD1, локализованном в регионе 16p13.3 и кодирующем белок полицистин-1 (в этом случае средний возраст развития терминальной почечной недостаточности составляет 54 года), 15 % случаев связаны с мутацией гена PKD2 в регионе 4q21 и кодирующем белок полицистин-2 (средний возраст развития терминальной почечной недостаточности составляет 74 года)[5][6].
Аутосомно-доминантная поликистозная болезнь почек в 90 % случаев наследуется от родителей, тогда как примерно в 10 % случаев является результатом спонтанных мутаций гена.
Сами кистозные структуры бывают 2 видов:
- Закрытые капсулы. Это замкнутые полости, которые не имеют связи с мочевыводящим протоком. Такая форма образований чаще определяется у новорожденных при аутосомно-рецессивном поликистозе почек.
- Открытые. Представляют собой выпячивания стенок канальцев, которые сообщаются с лоханками. При возникновении таких форм, более характерных для аутосомно-доминантного поликистоза почек, выделительная функция органа сохраняется на протяжении длительного времени.[7]
Помимо этого, кисты в почках могут образовываться и при других заболеваниях, однако в этом случае их развитие связано с мутацией других генов.
Причины[править | править код]
Поликистоз почек передается по наследству, в равной степени встречается и у мужчин, и у женщин[8].
Механизмы развития[править | править код]
Аутосомно-доминантная и аутосомно-рецессивная поликистозная болезнь почек относится к цилиопатиям — группе заболеваний, для которых характерно нарушение нормальной работы ресничек на поверхности ряда клеток, за счет которых обеспечивается «прием» сигналов из внеклеточной среды. Белки полицистин-1, полицистин-2 и фиброцистин входят в состав первичных ресничек на поверхности клеток млекопитающих. В клетках эпителия почечных канальцев первичные реснички располагаются со стороны просвета почечных канальцев, и предполагается, что это обеспечивает их сенсорную функцию — чувствительность к току мочи.
В результате неправильного восприятия сигналов из-за нарушенной работы первичных ресничек в клетках почечного эпителия происходит накопление циклического аденозинмонофосфата, на снижение уровня которого направлено действие ряда экспериментальных методик лечения поликистоза почек.
На макроуровне для поликистоза характерно наличие множественных кист (отсюда название: поли- + киста + -оз) в обеих почках. Кисты образуются из-за повышенной пролиферации и дифференцировки эпителия канальцев нефрона. В результате вместо нормальных почечных канальцев образуются наполненные жидкостью пузырьки — кисты, что приводит к значительному увеличению объёма почек (вес почки больного может достигать 35 кг). Кисты в почке больного возникают фокально, не более чем в 2-5 % нефронов, но из-за увеличения объёма кист происходит сдавление соседних здоровых нефронов, и постепенно почка теряет фильтрующую функцию.
Кроме того, поскольку первичные реснички имеются в клетках других органов, при поликистозе почек также часто развиваются кисты в печени, поджелудочной железе, в сосудах головного мозга.
Лечение[править | править код]
Необходимо отметить, что течение поликистоза почек зависит не только от дефектного гена, но и от многих других факторов (в частности, хороший контроль артериального давления и своевременное лечение сопутствующего пиелонефрита позволяет замедлить прогрессирование хронической почечной недостаточности). Роль артериального давления и других факторов в прогрессировании поликистоза почек должно выявить проходящее в настоящее время исследование HALT PKD[9]
В настоящее время в клинической практике не доказана эффективность и безопасность ни одного из существующих лекарств на коррекцию первичных механизмов развития поликистоза почек. Лечение состоит в симптоматической терапии, направленной на нормализацию артериального давления, лечение сопутствующего пиелонефрита, ренопротективной терапии для замедления развития хронической почечной недостаточности.
При развитии терминальной хронической почечной недостаточности пациенту необходима заместительная почечная терапия — гемодиализ, перитонеальный диализ, трансплантация почек.
Образ жизни при поликистозе почек[править | править код]
Грамотный рацион питания и приема жидкостей позволяет существенно снизить темпы прогрессирования поликистоза и сопутствующих ему заболеваний (в том числе, отказ почек, гипертония и проч.). Рекомендации в плане рациона направлены в основном на поддержание низкого уровня артериального давления.
Больному поликистозом следует придерживаться следующих ограничений в плане питания[10][11]:
1. Ограничение поступления солей натрия (прежде всего, поваренной соли, которая способствует повышению артериального давления и создает дополнительную нагрузку на почки).
2. Уменьшение в рационе жирной (холестерин) и белковой пищи.
3. Исключение из рациона продуктов, содержащих кофеин (кофе, чай, шоколад и т. п.), который существенно ускоряет рост кист.
4. Необходимо достаточное потребление жидкости.
Кроме этих мер, специалисты рекомендуют отказаться от курения, приема гормональных препаратов, лекарств, оказывающих токсическое действие на почки. Необходимо поддерживать низкий уровень артериального давления (за счет блокады системы ренин — ангиотензин — альдостерон): как правило, в пределах 130/90, в некоторых случаях рекомендуют — не ниже 120/80.
Экспериментальные методы лечения[править | править код]
Хотя апробированного в клинике эффективного лечения нет, во всем мире активно ведутся поиски препаратов, направленных на замедление роста кист и специфическое для поликистоза торможение развития почечной недостаточности.
В клинических исследованиях с участием людей активно исследуются препараты, действие которых связано с уменьшением накопления в клетках циклического аденозинмонофосфата (аналоги соматостатина, антагонисты V2-рецепторов вазопрессина, ингибиторы mTOR).
Имеется также ряд препаратов, которые исследовались только на лабораторных животных. В частности, О. Ю. Бескровной на лабораторных мышах показана возможность блокировать развитие ПБП ингибированием циклин-зависимых киназ (то есть остановкой пролиферации эпителия кисты)[6], а также ингибированием синтеза гликозилцерамида[12].
Примечания[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Статья о поликистозной дисплазии почек Архивная копия от 3 марта 2011 на Wayback Machine в Малой медицинской энциклопедии
- ↑ Справочник-путеводитель практикующего врача.
2000 болезней от А до Я (под ред. Денисова И. Н., Улумбекова Э. Г.), М.: ГЭОТАР-МЕД, 2003. Статья Болезнь поликистозная почек Архивная копия от 1 марта 2009 на Wayback Machine. ISBN 5-9231-0240-4 - ↑ Torres et al. Autosomal dominant polycystic kidney disease. The Lancet — vol. 369 (9569), pp. 1287—1301. 2007
- ↑ Beskrovnaya O, Bukanov N. (2008) Polycystic kidney diseases: from molecular discoveries to targeted therapeutic strategies. Cell Mol Life Sci 65, 605-19.
- ↑ 1 2 Nikolay O. Bukanov, Laurie A. Smith, Katherine W. Klinger, Steven R. Ledbetter and Oxana Ibraghimov-Beskrovnaya (2006) Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 444, 949—952.
- ↑ Поликистоз почек: что это такое, причины, симптомы и лечение (рус.), ikista.ru (4 февраля 2018). Дата обращения 17 сентября 2018.
- ↑ Медицинская энциклопедия. — Астрель. — С. 382.
- ↑ Polycystic Kidney Disease Treatment Network (недоступная ссылка). Дата обращения 11 декабря 2010. Архивировано 1 июня 2010 года.
- ↑ Ronald D. Perrone, What every Patient should know about PKD (недоступная ссылка). Дата обращения 7 января 2014. Архивировано 7 января 2014 года.
- ↑ • Vitalfood.ru: Диета при поликистозе почек
- ↑ Thomas A Natoli, Laurie A Smith, Kelly A Rogers, Bing Wang, Svetlana Komarnitsky, Yeva Budman, Alexei Belenky, Nikolay O Bukanov, William R Dackowski, Hervé Husson, Ryan J Russo, James A Shayman, Steven R Ledbetter, John P Leonard & Oxana Ibraghimov-Beskrovnaya (2010) Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models.Nature Medicine 16, 788—792
Ссылки[править | править код]
- Лекции, статьи и новости по нефрологии
- PKD Foundation (англ.)
- Ronald D. Perrone. What every Patient should know about PKD. — June 22, 2007. — С. 9.
Источник
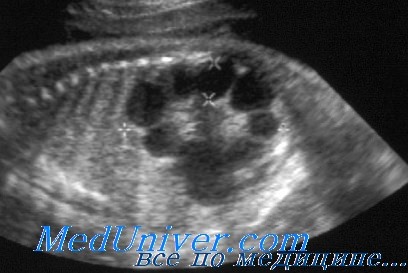
Поликистоз почек аутосомно-доминантный (АДПКП): причины, диагностика, лечениеЭтиология и встречаемость поликистоза почек. Аутосомно-доминантный поликистоз почек (АДПКП) (MIM №173900) — генетически разнородное заболевание. Приблизительно у 85% пациентов поликистоз I типа вызван мутациями в гене PKD1; большинство остальных имеют поликистоз II типа вследствие мутаций в гене PKD2. Несколько семей не показывают сцепления с этим локусами, что позволяет предполагать наличие, по крайней мере, еще одного дополнительного, пока неопознанного локуса. Аутосомно-доминантный поликистоз почек (АДПКП) — одно из наиболее частых генетических заболеваний, имеет распространенность от 1 на 300 до 1 на 1000 во всех изученных этнических группах. В США заболевание составляет 8-10% терминальной почечной недостаточности. Патогенез аутосомно-доминантного поликистоза почек (АДПКП)Ген PKD1 кодирует полицистин-1, трансмембранный рецептороподобный белок неизвестной функции. Ген PKD2 кодирует полицистин-2, мембранный белок, гомологичный натриевому и кальциевому а1 каналам. Полицистин-1 и полицистин-2 взаимодействуют как часть гетеромногомерного комплекса. Образование кист при аутосомно-доминантном поликистозе почек (АДПКП), как оказалось, соответствует «двухударному» механизму, наблюдающемуся при мутациях генов-супрессоров опухолевого роста и новообразованиях; т.е. для формирования кист должны утратить функцию оба аллеля гена PKD1 или PKD2. Механизм функциональной причины образования кисты при утрате функций полицистинов до конца не определен, но включает неправильное расположение белков на поверхности клетки, в норме ограниченное базолатеральной или эпителиальной поверхностью формирующихся тубулярных клеток почки.
Фенотип и развитие аутосомно-доминантного поликистоза почек (АДПКП)Аутосомно-доминантный поликистоз почек (АДПКП) может обнаруживаться в любом возрасте, но симптомы или признаки чаще появляются на третьем-четвертом десятилетиях жизни. У пациентов появляются инфекции мочевых путей, гематурия, нарушения уродинамики (сгустки или камни почек), ноктурия, кровоизлияния в кисты или жалобы на боли в боку, вызванные увеличением размеров расширенных почек. Артериальная гипертензия бывает у 20-30% детей и почти у 75% взрослых с аутосомно-доминантным поликистозом почек. Гипертония — вторичный эффект внутрипочечной ишемии и активизации системы ренин-ангиотензин. Почти половина больных к 60 годам жизни имеет почечную недостаточность. Гипертония, повторные инфекции мочевых путей, мужской пол и возраст начала клинических проявлений — наиболее важные факторы, указывающие на раннее развитие почечной недостаточности. Приблизительно 43% пациентов с началом болезни вскоре после рождения умирают от почечной недостаточности в течение первого года жизни; у остальных к 30 годам жизни формируется терминальная почечная недостаточность, гипертония или и то, и другое вместе. Аутосомно-доминантный поликистоз почек (АДПКП) демонстрирует как межсемейную, так и внутрисемейную изменчивость по возрасту начала и тяжести. Часть междусемейной изменчивости вторична относительно локусной гетерогенности, поскольку пациенты с поликистозом II типа имеют более мягкие проявления болезни, чем пациенты с I типом болезни. Внутрисемейная изменчивость, как оказалось, вызвана совместным влиянием среды и генетического фона, поскольку изменчивость более выражена между поколениями, чем среди сибсов. Кроме кист почек, у пациентов с аутосомно-доминантным поликистозом почек образуются кисты в печени, поджелудочной железе, яичниках и селезенке, а также внутричерепные аневризмы, пролапс митрального клапана и дивертикулы толстой кишки. Кисты печени часто встречаются как при АДПКП-1, так и при АДПКП-2, тогда как кисты поджелудочной железы обычно наблюдают при АДПКП-1. Внутричерепные мешотчатые аневризмы выявляют у 5-10% больных АДПКП; тем не менее риск развития аневризм неодинаков для всех пациентов, поскольку они имеют семейное накопление. Больные аутосомно-доминантным поликистозом почек имеют повышенный риск недостаточности аортального и трехстворчатого клапанов, а пролапс митрального клапана выявляют у 25% больных. Дивертикулы толстого кишечника — наиболее частая внепочечная аномалия; при этом разрыв дивертикула при аутосомно-доминантном поликистозе почек более вероятен, чем при дивертикулах, наблюдаемых в общей популяции. Особенности фенотипических проявлений аутосомно-доминантного поликистоза почек (АДПКП):
Лечение аутосомно-доминантного поликистоза почек (АДПКП)В основном аутосомно-доминантный поликистоз почек диагностируют по семейному анамнезу и УЗИ почек. Обнаружение почечных кист при УЗИ увеличивается с возрастом, так что 80-90% пациентов имеют обнаруживаемые кисты к 20 годам и почти 100% — к 30 годам жизни. Если это необходимо для пренатальной диагностики или идентификации родственника-донора почки, диагноз можно подтвердить анализом сцепления или прямым обнаружением мутации, или, в некоторых семьях, и тем, и другим. Оказание помощи и лечение больных аутосомно-доминантным поликистозом почек нацелено на задержку развития почечной недостаточности и коррекцию симптомов. Гипертонию и инфекции мочевых путей следует энергично лечить, чтобы сохранить функцию почек. Боли, вызванные увеличением почек, уменьшаются при дренировании и склерозировании кист. Риски наследования аутосомно-доминантного поликистоза почекПриблизительно 90% пациентов имеют аутосомно-доминантный поликистоз почек в семейном анамнезе; только 10% аутосомно-доминантного поликистоза почек возникает вследствие новых мутаций в генах PKD1 или PKD2. Родители с аутосомно-доминантным поликистозом почек имеют 50% риск родить больного ребенка при каждой беременности. Если родители имели ребенка с внутриутробным началом болезни, риск родить другого сильно пораженного ребенка составляет приблизительно 25%. Тем не менее точно предсказать тяжесть проявлений болезни невозможно из-за варьирующей экспрессивности. Для семей, в которых известна мутация или возможен анализ сцепления, риск повторения может быть изменен при анализе ДНК плода. Сибсы и родители пациентов с аутосомно-доминантным поликистозом почек также имеют повышенный риск болезни. Для обследования членов семьи рекомендуемый метод — ультрасонография почек. Пример аутосомно-доминантного поликистоза почек (АДПКП). П.Д., 35-летний мужчина с пролапсом митрального клапана в анамнезе, поступает в местное отделение неотложной помощи с выраженными болями в боку и гематурией. Четыре месяца тому назад у него появились эпизодические боли в боку. УЗИ почек выявило нефролитиаз и множественные кисты почек, характерные для поликистоза почек. Данные его клинического осмотра в норме, за исключением систолического шума, соответствующего пролапсу митрального клапана, небольшой артериальной гипертензии и легкого повышения концентрации креатинина в сыворотке. Его отец и сестра умерли от прорыва внутричерепных аневризм, а сын умер в 1 год от поликистоза почек. После смерти сына врачи предлагали обследовать его и его жену на наличие поликистоза почек; однако родители решили не обследоваться из-за чувства вины и горя, вызванных смертью сына. Пациенту начато лечение камней в почках. Во время лечения нефролог сообщил больному, что у него аутосомно-доминантный поликистоз почек (АДПКП). — Также рекомендуем «Синдром Прадера-Вилли: причины, диагностика, лечение» Оглавление темы «Врожденные болезни»:
|
Источник