Врожденная расщелина губы и неба синдромы

Частота, причины врожденной расщелины верхней губы и неба у ребенкаРасщелины верхней губы и неба являются наиболее распространенными врожденными пороками головы и шеи. Расщепление верхней губы образуется при неполном слиянии верхнечелюстного и медиального отростков, твердого неба— при неслиянии латеральных небных выступов. Расщепление губы и расщепление неба могут встречаться как вместе, так и изолированно, возможно их сочетание с другими аномалиями головы и шеи. Диагностируется данный порок развития сразу после рождения, помимо очевидного косметического дефекта, в раннем возрасте он ведет к проблемам с дыханием и с приемом пищи. Рождение ребенка с расщелиной губы и неба может стать тяжелым событием для родителей. В этой главе обсуждаются генетические основы, анатомия и лечение данной аномалии развития. Также упомянуты проблемы нормального формирования речи. При написании этой главы мы не стремились заменить существующие подробные руководства, а скорее представить обзор методов лечения и ухода за такими больными. Частота и причины врожденных расщелин верхней губы и неба (эпидемиология и генетика). Расщепление губы и неба является второй по частоте встречаемости врожденной аномалией (после вальгусной деформации стопы). Как правило, расщепление верхней губы, как сочетающееся, так и изолированное от расщепления твердого неба, и изолированное расщепление неба считаются двумя разными аномалиями. В США суммарная встречаемость данных пороков развития составляет около одного случая на 600 живорождений. Согласно результатам исследования заболеваний зубов и лицевой области, проведенному Национальными институтами здравоохранения в 2006 году, расщепление верхней губы в сочетании или без с расщеплением твердого неба встречается с частотой один случай на 940 рождений, изолированное расщепление твердого неба — один случай на 1500 рождений. У мальчиков расщелина верхней губы встречается в два раза чаще, чем у девочек. Изолированное расщепление неба, напротив, встречается в два раза чаще у девочек. Наиболее часто заболевание встречается у представителей коренных американских народностей, затем идут представители монголоидной расы, европеоидной, и, наконец, афроамериканцы.
Расщепление верхней губы и расщепление неба могут быть как частью какого-то генетического синдрома, так и представлять собой несиндромальную форму. Рекомендуется проведение генетического консультирования, т. к. всегда имеется риск того, что явная расщелина твердой губы или неба является частью какого-либо генетического синдрома. Специфические генетические синдромы развиваются либо в результате изменений в каком-либо одном гене (аутосомно-доминантных, аутосомно-рецессивных, сцепленных), либо в результате хромосомных нарушений (делеция, транслокация, трисомия). На частоту встречаемости расщепления губы и неба влияет множество факторов, как генетических, так и негенетических. Известными факторами риска со стороны матери являются сахарный диабет, гестационный диабет, дефицит фолиевой кислоты, воздействие препаратов (этанол, фенитоин, талидомид), факторов окружающей среды (например, табачного дыма). Была обнаружена связь между развитием несиндромальных расщелин и геном, отвечающим за экспрессию трансформирующего фактора роста альфа (TGF-a). В настоящее время он является объектом многих исследований, направленных на выяснение этиологии данного порока развития. Синдромы, сопровождающие расщепление верхней губы и/или неба, перечислены в таблице ниже. Чаще всего они развиваются в результате нарушений в каком-либо генетическом локусе. Знание о наличии наследственного синдрома помогает родителям своевременно получить доступ к программам реабилитации и вовремя начать коррекцию сопутствующих нарушений развития, если таковые возникнут.
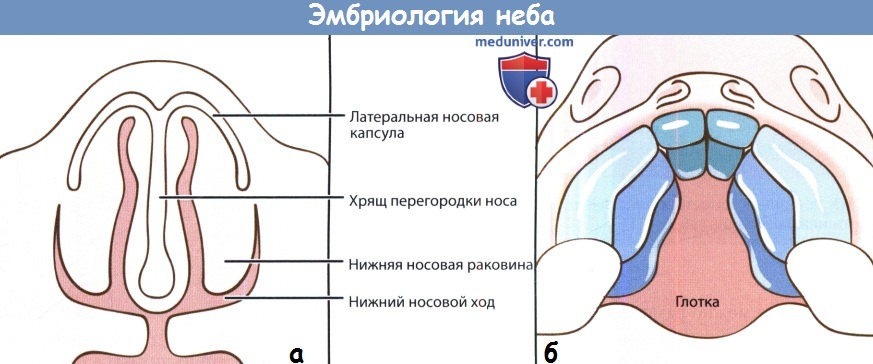
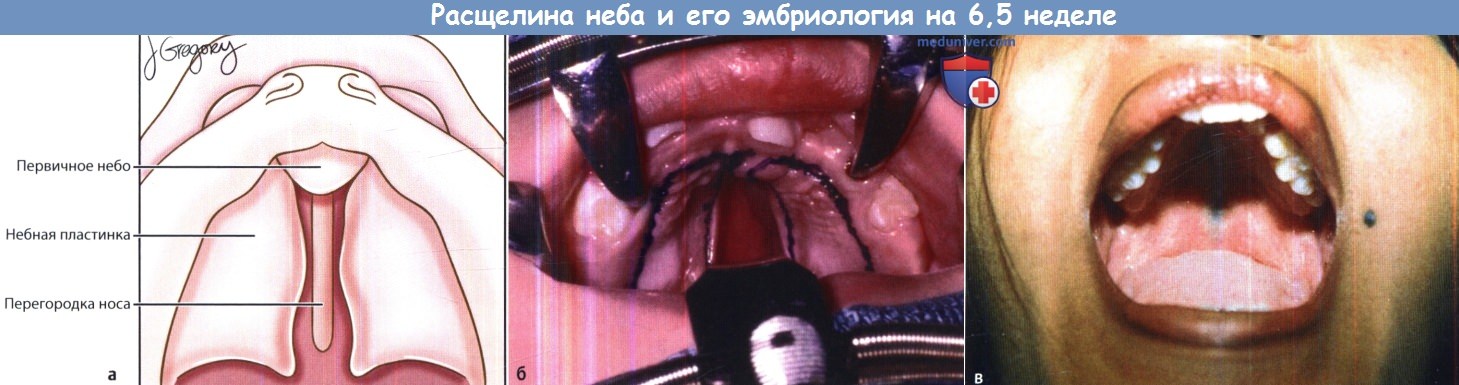
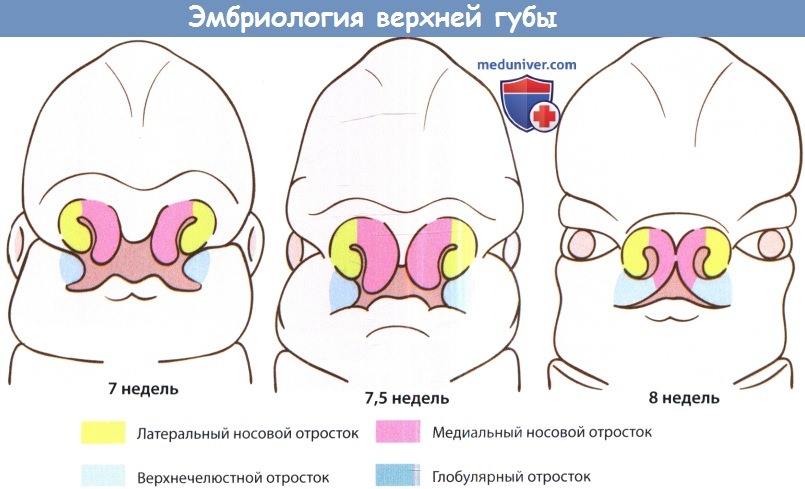
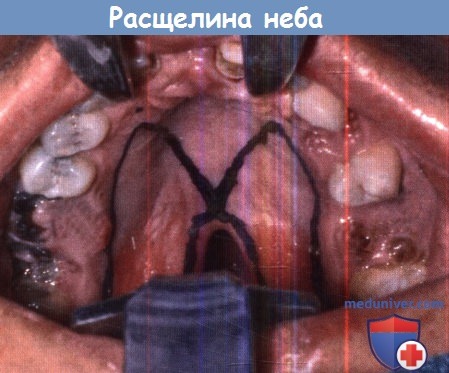
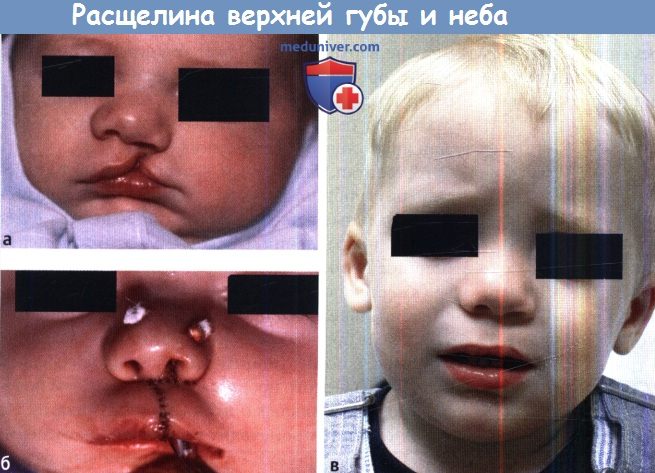
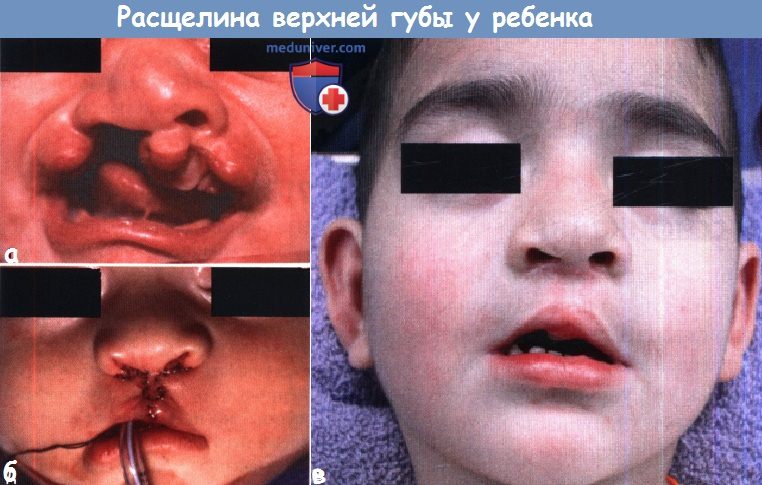
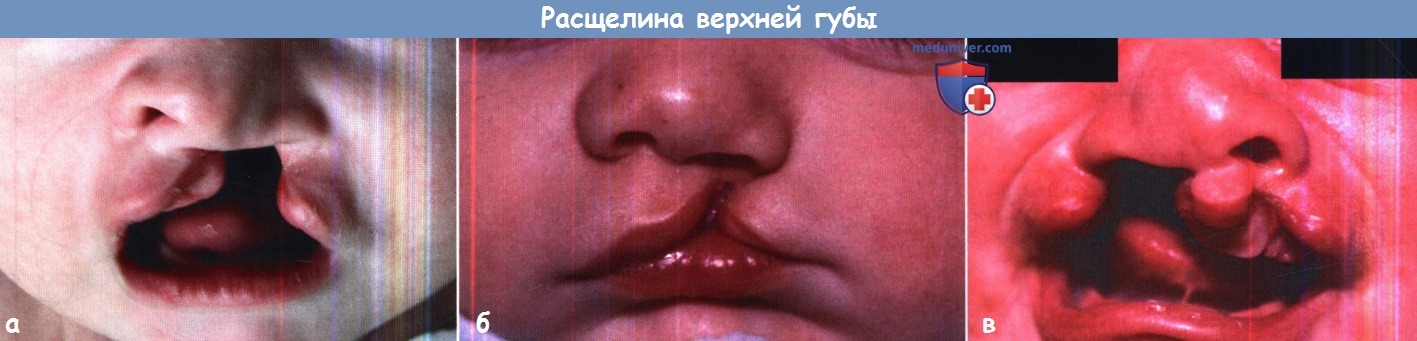
Эмбриология. Изолированная расщелина неба и расщелина верхней губы с сочетанием либо без сочетания с расщелиной неба считаются разными заболеваниями не только из-за разницы в частоте их встречаемости, но и из-за того, что они возникают на различных этапах эмбрионального развития. Формирование губ и неба начинается достаточно рано, еще в первом триместре беременности, состоит из двух связанных этапов. Первый начинается на 4-5 неделе внутриутробной жизни, во время него формируются губы, нос и первичное небо (премаксилла или костная пластинка с альвеолярными дугами и четырьмя передними резцами, расположенными кпереди от резцового отверстия). Второй этап, начинающийся на 8-9 неделе, представляет собой закрытие вторичного неба (путем слияния двух боковых небных выступов, содержащих мягкое небо и все остальные зубы). Эмбриология расщепления губы. На 4-5 неделях внутриутробной жизни происходит пролиферация эктодермы и мезодермы фронтоносового и латеральных верхнечелюстных отростков. Из лобноносового отростка в дальнейшем формируются следующие три структуры: губной желобок, центральный альвеолярный отросток, содержащий четыре передних резца (передний небный сегмент) и участок твердого неба, расположенный кпереди от резцового отверстия (задний небный сегмент). Этот процесс завершается на шестой неделе слиянием двух латеральных верхнечелюстных отростков с центральным лобно-носовым отростком. Лобно-носовой отросток образуется при дифференцировке эпителия обонятельной плакоды, одновременно с ним формируется завиток крыла носа. Слияние начинается у преддверия носа, формируется характерная форма ноздрей, а затем спускается книзу, к губам. Одновременно латеральные небные пластинки сливаются с центральной премаксиллой, соединяя альвеолярные дуги. В основе современной теории эмбрионального развития верхней губы и неба лежит идея о том, что в результате краевого контакта отростков происходит слияние и дифференциальная резорбция эпителиальных клеток, ремоделирование и слияние мезодермы с последующей дифференцировкой в костную, мышечную, слизистую и кожные ткани. Направлением, в котором происходит слияние трех отростков, объясняется большая выраженность расщепления губы и неба. Расщепление верхней губы может быть односторонним или двусторонним, полным (вплоть до носа) и неполным (от красной каймы губ до носа). Альвеолярные отростки могут быть как соединены (в норме), так и иметь ту или иную степень расхождения. Второй этап, или небный, завершается на 10 неделе внутриутробного развития. Увеличиваясь в размерах, латеральные небные пластинки смещаются в медиальном направлении. Поскольку к этому моменту первичное небо уже сформировалось на шестой неделе (вместе с верхней губой), вторичное небо (кзади и латеральнее резцового отверстия) претерпевает аналогичные процессы резорбции эпителия и слияния мезодермы, с последующим слиянием вдоль средней линии. В начале этого процесса зачаток языка расположен между латеральными небными пластинками. По мере их слияния происходит постепенное опущение языка. Спереди процесс ограничен резцовым отверстием, сзади — малым язычком. Этот факт объясняет наличие различных форм расщепления твердого неба: от расщелины язычка до полного расхождения вторичного неба (расщелина продолжается до резцового отверстия).
— Также рекомендуем «Классификация расщелины неба и верхней губы у ребенка» Оглавление темы «Болезни губы, неба, гортани у детей»:
|


Источник
Группа синдромов с неустановленным типом наследования была представлена синдромом Корнелии де Ланге, синдромом Гольденара, ассоциацией CHARGE и спорадическими случаями аномалада Пьера-Робена и синдрома Коффина-Сириса.
Как видно из табл. 2, наиболее частой формой мономутантных синдромов у пациентов с ВРГН явился аномалад Пьера-Робена, на долю которого пришлось 23,3 % (17 пациентов) всех случаев ВРН, входящих в синдромы. Все дети с аномаладом Пьера-Робена имели сочетание микрогении, глоссоптоза и расщелины неба.

Рис. 2. Распределение наследственных синдромов среди пациентов с ВРГН: 1 — АР; 2 — РГ; 3 — РГН левосторонняя; 4 — РГН правосторонняя; 5 — РГН двусторонняя; 6 — РН; 7 — абсолютное количество больных с ВРГН
Структура и частота сочетаний аномалада Пьера-Робена с другими пороками развития представлена в табл. 3. В 24 % случаев (4 пациента) аномалад Пьера-Робена явился составной частью синдромов множественных пороков развития: черепно-глазо-зубного синдрома (2 пациента), оро-фацио-дигитального (1 пациент) и синдрома эктодермальной дисплазии (1 пациент). Эти данные согласуются с литературными, где также указывается, что приблизительно в 25 % случаев аномалад Робена является составной частью синдромов множественных пороков.
Таблица 3. Структура и частота сочетаний аномалада Пьера-Робена с другими пороками развития и патологическими состояниями

Во всех случаях, когда аномалад Пьера-Робена являлся составной частью ЧГЗ-синдрома, отмечались: краниосиностоз, аномальная форма зубов, брахидактилия и аномалии глаз (птоз, страбизм, прогрессирующая миопия). Больной с синдромом Мора имел полидактилию кисти, синдактилию стоп на фоне гипосомии. У пациента с синдромом эктодермальной дисплазии наблюдались: сухая тонкая кожа, редкие тонкие волосы, гипоплазия и дистрофия ногтей и зубов, микроретрогения, аномалии глаз (блефарофимоз, эпикант, страбизм), атрезия слезных точек и слезных каналов.
У 1 больного аномалад Пьера-Робена проявился на фоне фетального алкогольного синдрома (пренатальная гипотрофия, микроцефалия с последующей умственной отсталостью, характерное лицо на фоне хронического алкоголизма матери).
Согласно литературным данным, у 36 % больных аномалад сочетается с другими пороками развития, в том числе с врожденным пороком сердца (ВПС), аномалиями скелета, глаз, ушных раковин, а также с умственной отсталостью. По нашим наблюдениям (табл. 3), четверть больных с аномаладом Пьера-Робена имели пороки развития опорно-двигательного аппарата — врожденную косолапость (2 пациента) и воронкообразную грудную клетку (2 пациента).
У всех без исключения пациентов наблюдались малые аномалии сердца, в 35 % случаев встречалось открытое овальное окно (6 человек). Большинство больных с аномаладом Робена имели дисплазии глаз, ушных раковин. Отмечены единичные случаи олигофрении, гипогонадизма. В 47 % случаев наблюдалась задержка психического развития (ЗПР), в 44 % — задержка физического развития (ЗФР).
Согласно табл. 2, с частотой 12-11 % встречались ЧГЗ-синдром и синдром эктодермальной дисплазии тип Рэппа-Ходжкина (по 9-8 пациентов, соответственно). Среди пациентов с ЧГЗ-синдромом только у троих отмечена изолированная РН, пятеро имели ВРГН, один пациент — атипичную двустороннюю косую расщелину лица. У всех больных с ЧГЗ-синдромом отмечалась асимметрия черепа вследствие краниосиностоза, аномальная форма зубов, аномалии глаз (птоз, страбизм, прогрессирующая миопия, атрофия зрительного нерва), аномалии ушных раковин, брахидактилия, иногда в сочетании с клинодактилией V.
Отмечалось асимметричное лицо с низким ростом волос на лбу, высокая спинка носа, гипертелоризм. В одном случае наблюдалось снижение слуха. С частотой 22 % синдром сочетался с аномалиями почек (подковообразная почка, неполное удвоение обеих почек) и с крипторхизмом (по 2 пациента). В 44 % (4 больных) отмечена ЗПР.
При синдроме эктодермальной гипогидротической дисплазии (тип Рэппа-Ходжкина) наблюдались: сухая тонкая кожа, редкие тонкие волосы, гипоплазия и дистрофия ногтей и зубов, запавшая переносица, микрогнатия, микростомия. 7 пациентов имели ВРГН и только один пациент имел изолированную РГ. В двух случаях отмечена гипоплазия гениталий. В единичных случаях встречались крипторхизм, синдактилия, сколиоз II ст., страбизм, ЗПР.
Синдром Смита-Лемли-Опица был выявлен у 5 пациентов (7 %). Все больные имели низкую массу и длину тела при рождении, микроцефалию с различными деформациями черепа (скафо- и долихоцефалию), узкий лоб, деформированные ушные раковины, микрогнатию, длинный фильтр и РН. Встречались птоз, эпикант, страбизм, сосковый гипертелоризм, крипторхизм, грыжи. Из аномалий конечностей отмечалась кожная синдактилия кистей и стоп. Со стороны внутренних органов наблюдались ВПС (дефект межжелудочковой перегородки), болезнь Гиршпрунга, дополнительная доля селезенки. У всех больных отмечалась умственная отсталость.
По 5,5 % пришлось на синдром эктодермальной дисплазии с синдактилией и EEC. В четырех случаях имел место синдром ВРГН с эктодермальной дисплазией и синдактилией, сопровождающийся олигофренией в стадии дебильности. Отмечались алопеция, дисплазия ногтей, гипоплазия эмали и раннее выпадение зубов, гипоплазия гениталий, микроблефарон (у 1 пациента).
При синдроме EEC отмечалась частичная синдактилия, РГН (3 пациента), изолированная РГ (1 пациент). Все пациенты имели проявления эктодермальной дисплазии: светлые, редкие, тонкие волосы, редкие брови и ресницы, сухую кожу, диспластический рост зубов. Отмечались гипоплазия ногтей, врожденный уретерогидронефроз, солитарная киста почки, добавочная доля селезенки. Умственное развитие всех больных соответствовало норме.
По 4 % пришлось на ГЗП-синдром, синдром Ван дер Вуда и синдром Корнелии де Ланге.
ГЗП-синдром встретился у 3 больных с левосторонней ВРГН. Больные имели микроофтальмию, микрокорнеа, колобому радужки, гипотелоризм, врожденную катаракту, узкие глазные щели, эпикант. Отмечались диспластический рост зубов, гипоплазия эмали, ранний кариес, синдактилия, клинодактилия V, брахицефалия. Двое больных имели врожденный вывих бедра, один проводящую глухоту. Психомоторное развитие детей соответствовало норме.
Все больные с синдромом Ван дер Вуда имели ямки на слизистой поверхности нижней губы (врожденный свищ), ВРГН.
Синдром Корнелии де Ланге наблюдался у 3 больных (4 %). При данном синдроме больные имели характерное лицо (синофриз, гипертрихоз), длинный фильтр, микрогению, микробрахицефалию, клинодактилию V. У всех больных помечались отставание в росте и глубокая задержка умственного развития. У двоих — судорожный синдром на фоне мышечного гипертонуса. Все пациенты имели ВПС (дефект межпредсердной перегородки, стеноз аортального клапана со стенозом легочной артерии, открытое овальное окно). Наблюдались: атрезия слезных точек, мультикистоз почки, крипторхизм.
С частотой 2,7 % каждый встречались синдромы Опица-Фриаса, Бикслера, Гольденара, Ваарденбурга, Коффина-Сириса, CHARGE-ассоциация, тригоноцефалия Опица (по 2 пациента каждый).
В двух случаях был диагностирован синдром Ваарденбурга. Лицевые аномалии включали телекант, широкую выступающую переносицу, прогению, микрогнатию, одностороннюю РГН. Отмечался частичный альбинизм, гетерохромия радужных оболочек, птоз.
Основными признаками синдрома Коффина-Сириса были грубые черты лица, гипоплазия V пальца и ногтей на пальцах стоп. Отмечались РН, односторонняя РГН, гипоплазия ногтей на пальцах рук, гипермобильность суставов, плоскостопие, порок сердца (дефект межжелудочковой перегородки с клапанным стенозом легочной артерии), крипторхизм, эктопия почки, глубокая умственная отсталость, незначительная микроцефалия, гидроцефалия. Редкие волосы на голове сочетались с генерализованным гирсутизмом.
Лицевые дисморфии при синдроме Опица-Фриаса включали гипертелоризм, уплощенную переносицу, узкие глазные щели, эпикант, аномальный разрез глаз, страбизм, микрогнатию, РГ, РГН. Гипоспадия была представлена в двух формах: венечной и гипоспадии полового члена. Отмечались открытое овальное окно, пиелоэктазия почки. Интеллект у больных был снижен.
У больной с синдромом Гольденара наблюдались: скрытая РН, правосторонняя гипоплазия нижней челюсти, аномальные ушные раковины с преаурикулярным выростом на стороне поражения, односторонний дермоид конъюнктивы справа, антимонголоидный разрез глаз с уменьшением глазной щели на стороне поражения, дефекты глазодвигательных мышц, воронкообразная деформация грудной клетки, аномалии позвоночника (клиновидный полупозвонок в грудном отделе, добавочное ребро, сколиоз), кривошея, умеренная задержка психического развития.
У другого больного с синдромом Гольденара отмечались: левосторонняя макростома, левосторонняя гипоплазия нижней и верхней челюсти, гипоплазия ушной раковины с преаурикулярными выростами на стороне поражения, атрезия наружного слухового прохода, глухота, антимонголоидный разрез глаз с уменьшением глазной щели на стороне поражения, дефекты глазодвигательных мышц, ЗПР.
Синдром Бикслера проявился у больных с односторонними ВРГН, одна из которых была атипичной. Больные имели гипертелоризм, левостороннюю гипоплазию дуги нижней челюсти, левостороннюю микроотию с атрезией наружного слухового канала, брахицефалию, задержку физического развития. Отмечались арахнодактилия, аневризма межпредсердной перегородки, пупочная грыжа со свищем, плосковалыусная деформация стоп, гипоплазия доли щитовидной железы.
У одного пациента с CHARGE-ассоциацией наблюдались врожденная левосторонняя колобома радужки и сосудов оболочки, атрезия левого носового хода, открытое овальное окно с выбуханием синуса Вальсальвы, ЗПМР, ЗФР, преаурикулярный вырост слева, левосторонняя РГ. Другой имел двустороннюю атрезию хоан, колобому радужки, ВПС (дефект межпредсердной перегородки), аномальные ушные раковины, глухоту, ЗПМР и ЗФР на фоне РН.
Синдром тригоноцефалии Опица у больной со щелевидной РН и у больного с атипичной РН проявился характерной формой черепа с расширением в затылочной и сужением в лобной части, лицевыми аномалиями в виде уплощенной с бороздкой спинки носа, монголоидного разреза глазных щелей, эпиканта, страбизма, аномалиями ушных раковин, повышенной эластичностью кожи, синдактилией стоп. Оба пациента имели ВПС (коарктация аорты с аномалией створок аортального клапана, дефект межжелудочковой перегородки) и ЗФР.
Более редкими оказались наследственные синдромы Крузона, Дубовица, оро-фацио-дигитальньш I типа. У больной с синдромом Крузона отмечались брахицефалия за счет краниосиностоза, гипертелоризм, экзофтальм, клювовидный нос, микрогнатия, мандибулярный прогнатизм, РН, олигофрения. Ребенок с синдромом Дубовица имел пренатальное и постнатальное отставание в физическом развитии, микроцефалию, лицевые аномалии (гипертелоризм, эпикант, широкую переносицу, левостороннюю РГН, микрогнатию), страбизм, левосторонний крипторхизм, олигофрению в стадии дебильности. Диагностическими признаками для оро-фацио-дигитального синдрома I типа явились множественные гиперплазированные уздечки языка, РГН, аномалии зубов, эпикант, микрогнатия, асимметричное укорочение пальцев стоп, ЗПР в легкой степени.
Из наследственных мономутантных синдромов, не предполагающих в своем составе ВРГН, были диагностированы: синдром Элерса — Данлоса (I, II, VI типов), синдром Нунана и синдром Горлина. У одной пациентки было установлено нарушение в системе половых хромосом — синдром Шерешевского-Тернера.
Определенный интерес представляли комбинации врожденных расщелин лица и неба с пороками развития внутренних органов, значительное место среди которых занимали врожденные пороки сердца, от тяжести которых во многом зависит жизненный прогноз. Частота подобного сочетания, по нашим наблюдениям, составила 6,6 % (20 пациентов).
Ассоциация расщелины лица и ВПС в составе синдромов множественных пороков развития нами была выявлена у 7 пациентов (табл. 4). ВРГН, сочетающаяся с ВПС, но не составляющая определенный синдром — у 13 больных. По литературным данным частота сочетания ВРГН и ВПС составляет 3,8-17,9 %.
Таблица 4. Наследственные болезни с ассоциацией ВРГН, ВПС и крупных сосудов

Один пациент имел ВРГН, сочетающуюся с врожденным пороком почек, но не составляющую определенный наследственный синдром.
Таким образом, по нашим наблюдениям, не менее чем у трети больных ВРГН входит в состав наследственных синдромов или сочетается с другими врожденными пороками развития (94 больных — 31,3 %). В подавляющем же большинстве случаев (69 %) ВРГН бывает единственной патологией у ребенка (либо сопровождается малозначимыми дисплазиями).
Семейная отягощенность пороком лица имела место в 13,3 % случаев, по данным литературных источников этот процент несколько выше — 19,57 %.
Заключение
1. Проведенное исследование установило, что в 31,3 % случаев ВРГН входит в состав множественных пороков развития, в том числе на долю синдромов наследственной этиологии приходится 26,7 %, что изменяет перспективы хирургической коррекции у этих больных и сроки возможного оперативного лечения.
2. Маркером вероятности проявления наследственной патологии является наличие у больных расщелины нёба.
3. Наиболее частой формой мономутантных синдромов у пациентов с ВРГН является аномалад Пьера-Робена, на долю которого пришлось 23,3 %.
4. Комбинация ВРГН с врожденным пороком сердца, от тяжести которого во многом зависит жизненный прогноз, составляет 6,6 %.
5. В 26,7 % случаев, когда ВРГН входит в состав наследственного синдрома, риск повторяемости расщелин лица для потомства оценивается как высокий — 25-50%. Поэтому все больные с ВРГН нуждаются в консультации врача-генетика для выявления генетических синдромов, отягощающих течение порока и прогноза потомства.
6. Учитывая высокую пораженность детей с ВРГН другими врожденными пороками, необходимо широкое внедрение метода дородового ультразвукового скрининга беременных из групп риска по рождению детей с ВРГН, т.к. пренатальная диагностика способствует, с одной стороны, сокращению рождаемости детей с тяжелыми пороками, с другой — более раннему и эффективному хирургическому лечению.
7. В случае выявления у плода ВРГН необходимо тщательное УЗИ с пристальным изучением анатомии лица, мозга, сердца и скелета.
8. Диагностика генетических синдромов имеет значение для определения прогноза жизни, показаний к хирургической коррекции порока, своевременному распознаванию и предупреждению осложнений послеоперационного периода.
Форофонтова В. Ю, Лебедькова С. Е., Мамедов Ад. А., Очнева Г. И., Скойбедо И. Е., Афуков И. В.
Опубликовал Константин Моканов
Источник