У моего ребенка синдром миллера дикера

Синдром Миллера-Дикера: причины, диагностика, лечениеЭтиология и встречаемость синдрома Миллера-Дикера. Синдром Миллера-Дикера (MDS, MIM №247200) -синдром протяженной делеции, вызываемый гемизиготной делецией 17р13.3; механизм, лежащий в основе повторных делеции участка 17р13.3, еще не объяснен, но, возможно (подобно другим синдромам микроделеции), он включает рекомбинацию между повторяющейся ДНК-последовательностью с небольшим числом копий. Синдром Миллера-Дикера — редкое заболевание неопределенной встречаемости во всех популяциях. Патогенез синдрома Миллера-ДикераВ регионе делеции синдрома Миллера-Дикера в 17р13.3 картировано более 50 генов, но только ген LIS1 (MIM №601545) связан со специфической фенотипической характеристикой синдрома; гемизиготность по LIS1 вызывает лиссэнцефалию. LIS1 кодирует мозговую изоформу некаталитической бета-субъединицы фактора активации ацетилгидролазы тромбоцитов (PAFAH). PAFAH — ингибитор миграции нейронов и также связывает и стабилизирует микрофибриллы. Предварительные наблюдения указывают, что PAFAH может играть роль в реорганизации микрофибрилл, необходимой для миграции нейронов. Тем не менее изолированная гаплонедостаточность LIS1 не вызывает других дисморфических признаков, характерных для синдрома Миллера-Дикера. Мутации в гене LIS1 вызывают изолированную лиссэнцефалию (MIM №607432), т.е. лиссэнцефалию без других дисморфии. Поскольку все пациенты с синдромом Миллера-Дикера имеют дисморфические черты лица, этот дисморфизм должен вызываться гаплонедостаточностью одного или нескольких других генов в делеции.
Фенотип и развитие синдрома Миллера-ДикераСимптоматика синдрома Миллера-Дикера включает дисгенезию мозга, мышечную гипотонию, задержку развития и лицевые дисморфии. Дисгенезия мозга характеризуется лиссэнцефалией I (полная агирия) или II типа (широко распространенная агирия с несколькими бороздами на лобном или затылочном полюсе), церебральной корой с четырьмя вместо шести слоев, гетеротопией серого вещества и истончением белого вещества. У некоторых больных также отмечают пороки развития сердца и омфалоцеле. Дети с синдромом Миллера-Дикера плохо едят и растут. Способность улыбаться, краткий визуальный контакт и неспецифические двигательные реакции — единственные способности, приобретаемые большинством пациентов. Кроме умственной отсталости, пораженные обычно страдают от опистотонуса, спастичности и судорог. Почти все они умирают к 2 годам жизни. Особенности фенотипических проявлений синдрома Миллера-Дикера:
Лечение синдрома Миллера-ДикераЧерты лица больных и обнаружение на МРТ лиссэнцефалии часто позволяет предположить диагноз синдрома Миллера-Дикера. Тем не менее для подтверждения диагноза необходимо обнаружить делецию 17р13.3 при хромосомном анализе или FISH с LIS1-специфическим зондом. Приблизительно 60% пациентов имеют видимую делецию критического региона синдрома Миллера-Дикера. Синдром Миллера-Дикера неизлечим; следовательно, помощь направлена на коррекцию симптомов и паллиативный уход. Почти всем детям необходимо лекAPCтвенное лечение судорог. Большинство пациентов получают питание через назогастральный или гастростомический зонд из-за проблем вскармливания и повторных аспирации. Риски наследования синдрома Миллера-ДикераУ 80% пациентов бывает вновь возникшая микроделеция 17р13.3, а 20% наследуют делецию от одного из родителей, несущего сбалансированную хромосомную перестройку. Из-за частоты, с которой деления наследуется от родителя со сбалансированной транслокацией, анализ кариотипа и FISH на LIS1 следует выполнять у обоих родителей. Родитель со сбалансированной транслокацией, захватывающей 17р13.3, имеет приблизительно один шанс из четырех иметь аномального живорожденного ребенка (с синдромом Миллера-Дикера или с дупликацией dupl7p) и приблизительно один шанс из пяти — прерывания беременности. В отличие от этого, если пациент имеет синдром Миллера-Дикера в результате новой делеции, родители имеют низкий риск для повторения синдрома у последующих детей. Хотя порок развития мозга при синдроме Миллера-Дикера вызван неполной миграцией нейронов в церебральную кору во время 3-4 месяцев гестации, лиссэнцефалию не обнаруживают при МРТ или ультрасонографии плода до конца беременности. Для пренатальной диагностики синдрома Миллера-Дикера необходимо обнаружение делеции 17р13.3 в ворсинах хориона или амнио-цитах плода. Пример синдрома Миллера-Дикера. Б.Б., мальчик 5 дней жизни, родившийся на 38-й нед гестации, переведен в палату интенсивной терапии отделения новорожденных из-за выраженной гипотонии и затруднений вскармливания. Ребенок родился от неосложненной беременности; УЗИ плода на 14-й нед гестации и материнский сывороточный скрининг на 16-й нед гестации соответствовали норме. Мальчик родился от спонтанных влагалищных родов; оценка по шкале Апгар была 8 баллов на 1-й мин и 9 баллов на 5-й мин жизни. В семейном анамнезе ребенка генетические, неврологические или врожденные заболевания отсутствуют. При клиническом осмотре выявлены мышечная гипотония и слегка дисморфиче-ские черты лица, включая сужение битемпорального расстояния, вдавленную переносицу, небольшой нос с вывернутыми ноздрями и микрогнатию. В остальном данные обследования в норме. Его анализы на электролиты сыворотки крови, метаболический скрининг и врожденные инфекции оказались в пределах нормы. Ультразвуковое сканирование мозга показало гипоплазию мозолистого тела, легкое расширение желудочков мозга и сглаженную кору. После дополнительного совещания группа генетиков рекомендовала хромосомный анализ, флюоресцентный анализ (FISH) гена LIS1 (расположенный в регионе 17р13.3) и МРТ мозга. МРТ показала уплотнение коры мозга, полную агирию, многочисленные церебральные гетеротопии, гипоплазию мозолистого тела, нормальный мозжечок и ствол мозга. Хромосомный анализ с G-окраской оказался нормальным (46,XY), но FISH показал деле-цию LIS1 в одной из хромосом 17. На основе этих результатов генетик объяснил родителям, что у ребенка синдром Миллера-Дикера. Родители отказались от лечебных мероприятий, кроме необходимых для комфорта ребенка, и он умер в 2-месячном возрасте. — Также рекомендуем «Миоклонус-эпилепсия MERRF: причины, диагностика, лечение» Оглавление темы «Наследственные болезни»:
|
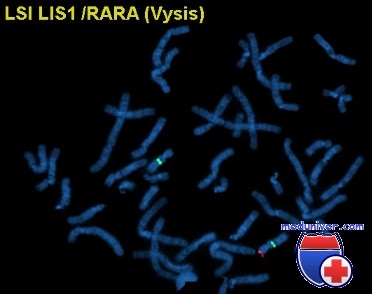
Источник
Синдром Миллера-Дикера — очень редкий синдром делеции 17p13.3. Этот синдром характеризуется лиссэнцефалией классического типа, отличительными чертами лица и другими врожденными пороками, которые встречаются только у некоторых лиц с этим синдромом.
Синдром Миллера-Дикера. Причины
Синдром Миллера-Дикера был открыт в 1983 году. Основной причиной развития этого синдрома является делеция части хромосомы 17p13.3. Почти 100% пациентов с диагнозом синдрома Миллера-Дикера имеют эту делецию. Размер делеции варьируется среди индивидуумов. Около 50% имеют микроскопически видимые делеции, а остальные имеют субмикроскопические делеции. Клинические особенности синдрома Миллера-Дикера связаны с потерей нескольких генов в хромосоме 17. Потеря гена PAFAH1B1, как уже известно исследователям, отвечает за развитие лиссэнцефалии. Потеря гена YWHAE увеличивает тяжесть лиссэнцефалии, другие гены, потеря которых приводит к появлению других клинических особенностей этого синдрома, еще не известны.
Синдром Миллера-Дикера обычно не наследуется от родителей. Делеция, которая является результатом семейной хромосомной транслокации, встречается лишь у около 12% больных. Чаще всего эта делеция происходит случайно в процессе формирования половых клеток или на ранней стадии беременности. Вероятность иметь другого ребенка с этим же синдромом очень низкая (если эта делеция не будет связана с транслокацией).
Синдром Миллера-Дикера. Симптомы и проявления
Лиссэнцефалия, при синдроме Миллера-Дикера, характеризуется агирией / агирией с фронтальной пахигирией и толстой корой больших полушарий головного мозга (~ на 3 мм толще нормальной). Дополнительные особенности включают в себя заднее расширение боковых желудочков и гипоплазию (недоразвитие) мозолистого тела.

Ребенок с синдромом Миллера-Дикера
Синдром Миллера-Дикера связан с умственной отсталостью, задержками физического развития, характерными лицевыми особенностями, необычной мышечной спастичностью, трудностями в кормлении, низким мышечным тонусом (гипотония) и судорогами. Задержки развития, как правило, тяжелые и большинство детей не смогут научиться сидеть или ходить самостоятельно. Их развитие будет постоянно оставаться на уровне развития здорового 3-6 месячного ребенка. Трудности с кормлением и глотанием являются общими и они могут осложняться аспирационной пневмонией. Низкий мышечный тонус (гипотония) является характерной особенностью этой аномалии и, как правило, она отмечается по всему телу. Приступы обычно начинаются в период между рождением и 6 месяцами. Окружность головы маленькая. Чем сильнее будут сглажены извилины, тем тяжелее будут симптомы и проявления. Характерные лицевые особенности включают: выпуклый лоб, затонувшее лицо, небольшой вздернутый нос, аномальные и низко расположенные уши, небольшие челюсти, толстая верхняя губа. Некоторые дети с этим синдромом растут медленнее, чем их здоровые сверстники. У некоторых детей отмечаются пороки развития сердца / почек и пупочная грыжа.
Синдром Миллера-Дикера. Диагностика
Беременность, осложненная плодом с синдромом Миллера-Дикера, может быть связана с многоводием (избыток амниотической жидкости), задержкой внутриутробного роста и снижением движений плода. Постановка диагноза и его подтверждение основывается на хромосомном анализе. Классическая лиссэнцефалия, как правило, становится видимой (при проведении МРТ) только с 28 недели беременности.
Синдром Миллера-Дикера. Лечение
Лечение только симптоматическое и зависит от тяжести симптомов. Контроль приступов имеет очень важное значение. Питательная трубка может быть использована для решения проблем с кормлением. Трудотерапия, физиотерапия и специальные образовательные программы могут помочь ребенку достичь потенциала на столько, на сколько это возможно.
Синдром Миллера-Дикера. Прогноз
Прогноз развития варьируется в зависимости от тяжести пороков развития головного мозга. Большинство детей умирают в течение первых нескольких лет жизни. Тем не менее, с улучшенным контролем приступов и с использованием питательной трубки, дети с этим синдромом живут дольше. Меньшинство пациентов с синдромом Миллера-Дикера доживают до раннего подросткового возраста. Аспирационная пневмония является наиболее распространенной причиной наступления смерти.
Источник
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке «Файлы работы» в формате PDF
Введение
В настоящее время значительно увеличилось рождение детей, страдающих тем или иным заболеванием с разной частотой появления в популяции, одни из которых могут не сильно сказываться на жизнедеятельности, а другие – могут резко ее ограничивать. Так, синдром, который я выбрала для изучения, обусловлен тяжелой задержкой психического и моторного развития ребенка с проявлением в большинстве случаев эпилептических приступов.
Цель данной работы состоит в изучении синдрома Миллера-Дикера.
Задачи:
1.Рассмотреть причины появления данного синдрома;2. Ознакомиться с классической лиссэнцефалией;
3. Изучить методы диагностики синдрома Миллера-Дикера;
4. Разобрать проявление синдрома на примере клинического случая
Синдром Миллера-Дикера
Синдром Миллера-Дикера (СМД) – редкое генетическое заболевание, обусловленное микроделециями в участке 17р13.3 и характеризующееся классической лиссэнцефалией в сочетании с лицевым дисморфизмом. Классическая лиссэнцефалия (тип 1, форма Бильшовского) проявляется малыми размерами мозга, утолщением коры, состоящей из 4 аномальных слоев с формированием только первичных и нескольких вторичных извилин. Возможны гипоплазия коры мозжечка, камптодактилия I-III пальцев стоп, врожденные пороки сердца, агенезия почек, крипторхизм, паховые грыжи [3, с.142].
Причиной синдрома считается микроделеция дистальной части короткого плеча 17 хромосомы (локус 17р13.3), которая встречается у подавляющего большинства пациентов. В 80 случаев возникает в результате делеции de novo, А в 20% — один из родителей является носителем сбалансированной мутации на хромосоме 17. В участке 17р13.3 был картирован ген LIS1, оказывающий прямое влияние на процессы нейрональной миграции в коре головного мозга и мозжечке. Потеря одного аллеля такого гена приводит к остановке и нарушению клеточной миграции с формированием лиссэнцефалии.
В некоторых случаях классической лиссэнцефалии возникает гемизиготная или гетерозиготная мутация в гене DCX, кодирующим белок даблкортин на Х хромосоме. Эта мутация может быть спонтанной или возникает в результате передачи по материнской линии. Женщины носительницы такого гена могут быть клинически здоровы или иметь подкорковую ленточную гетеротопию [4, с.61].
Классическая лиссэнцефалия – тяжёлая мальформация головного мозга человека, при которой поверхность больших полушарий сглажена; морфологически определяются первичные и единичные вторичные извилины; кора утолщена и состоит из 4 примитивных слоёв: молекулярный, поверхностный клеточный, губчатый и глубокий клеточный. Большинство нейронов в обоих клеточных слоях ориентированы аномально – косо или апикальными дендритами вниз. Глубокий клеточный слой состоит из эктопированных нейронов, остановившихся в процессе внутриутробной миграции из герминальных слоёв на 12 неделе гестации.
В общей популяции распространённость заболевания составляет 1-4 случаев на 100 тысяч. У пациентов с классической лиссэнцефалией частота встречаемости СМД достигает 30%. Синдром Миллера-Дикера проявляется классической лиссэнцефалией в сочетании с краниофациальными аномалиями. Для больных СМД характерно лиссэнцефалия 1-2 степени: тотальная агирия или агирия с участками пахигирии в лобно-височных областях. Больные имеют характерный внешний вид: высокий выступающий лоб, впалые виски, короткий нос с широкой переносицей и вздёрнутым кончиком, маленькая нижняя челюсть, тонкая выступающая кпереди верхняя губа. Другие нарушения встречаются несколько реже: эпикант, гипертелоризм, низкопосаженные ротированные назад уши. При рождении окружность головы может быть нормальной или уменьшенной, но в дальнейшем рост головы замедляется, поэтому у пациентов старше года – микроцефалия.
В пренатальном периоде наблюдается задержка внутриутробного развития и снижения двигательной активности плода. Беременность протекает на фоне многоводья. У новорожденных констатируется генерализованная гипотония, которая сменяется нарастающей спастичностью и тетрапарезом с позой опистотонуса. Для всех пациентов характерна тяжелая задержка моторного и психического развития. В большинстве случаев из приобретённых навыков у детей отмечается короткие и непостоянные эпизоды слежения глазами и улыбка, в редких случаях – контроль головы и способность переворачиваться на бок [4, с.62].
Эпилептические приступы – частный симптом при синдромах Ангельмана и Миллера-Дикера, а также нередко встречается при трисомии 21 хромосомы или при синдроме фрагильной Х-хромосомы [5, с.38]. Наблюдается более чем у 90% больных, проявляются инфантильными спазмами, миоклоническими, генерализованными тоническими спазмами, простыми и сложными фокальными, вторично-генерализованными, атипичными абсансами [2, с.35]. В 75% случаев приступы начинаются на первом полугодии жизни, у 50% пациентов – в первые три месяца. Типично начало заболевания с массивных билатеральных миоклонических приступов или инфантильных спазмов без типичной гипсоретмии на ЭЭГ. На первых месяцах жизни инфантильным спазмам могут предшествовать тонические приступы или эпизоды иктальных апноэ. У больных старше одного года наиболее частым типом приступа является короткие генерализованные тонические спазмы, нередко возникающие серийно. В старшем возрасте характерна комбинация фокальных и псевдогенерализованных приступов. Констатируется атипичные абсанции, миоклонические пароксизмы, а так же фокальные моторные и аутомоторные, вторично-генерализованные тонико- клонические приступы. Приступы резистентны к терапии АЭП.
Из экстроневральных нарушения часто встречаются врождённые пороки сердца, пороки органов мочеполовой системы, нарушения закладки конечностей и позвоночного столба [4, с.62].
При ЭЭГ исследовании у больных СМД практически отсутствует пространственная организация основной активности, а также ЭЭГ различия сна и бодрствования. Отличительным ЭЭГ признаком в межприступном периоде является диффузная быстрая активность с амплитудой до 100 мкВ и акцентом в лобно-центральных отведениях. Быстрая активность появляется после 6 месяцев и комбинируется с дельта волнами. У больных после полутора лет диффузная быстрая активность постепенно становится доминирующей. Она не блокируется при записи с открытыми глазами и слабо модифицируется во сне.
Основным методом верификации лиссэнцефалии при СМД является МРТ. Главный признак лиссэнцефалии при МРТ исследовании – изображение коры головного мозга в виде песочный часов. Кора при нейровизуализации напоминает мозг плода 13 недели гестации. Поверхность полушария почти полностью гладкая, за исключением нескольких борозд на нижней и боковой поверхности мозга. Характерно утолщение коры до 20мм. При LIS1, классическом генотипе СМД, морфологические изменения максимально выражены в теменно-затылочных отделах коры; при генотипе DCX – лобных и частично височных. При мутации этого гена выражена гипоплозия червя мозжечка. В стволе наблюдается эктопия ядер олив, гипоплазия пирамид. У 40% больных СМД обнаруживаются церебральные кальцификаты, локализованные по средней линии, которые отсутствуют при изолированной лиссэнцефалии. При лиссэнцефалии, обусловленной цитомегаловирусной инфекцией, могут обнаруживаться перивентрикулярные кальцификаты, которые не имеют отношения к СМД [4, с.63].
Клинический случай синдрома
Больная К.Д., 3 года. В возрасте ребенка 12 месяцев семья впервые обратилась за консультацией в Центр Детской Неврологии и Эпилепсии. Жалобы на частые эпилептические приступы и тяжелую задержку психо-моторного развития.
Из анамнеза заболевания известно, что в 2,5 мес. девочка была госпитализирована в отделение реанимации в связи с внезапно возникшей в ночное время серией эпизодов следующего характера: «сжималась», наблюдалось тоническое напряжение тела с клоническими подергиваниями дистальных отделов конечностей. Приступы протекали серийно, с потерей сознания; длительность каждого эпизода около 2 минут. Был назначен депакин хроно 300 в дозе 300мг/сут, на фоне приема которого отмечалось урежение приступов. В возрасте 6 мес появились эпизоды подергивания век, заведение глазных яблок вверх, нередко сопровождающиеся закидыванием головы назад. С частотой до 5 раз/сутки отмечались приступы, начинающиеся с кивка с последующим тоническим напряжением правой руки. В возрасте 8 мес был назначен синактен-депо, и приступы не отмечались в течение 3 месяцев. На момент первичной консультации у ребенка наблюдались симметричные тонические экстензорные приступы по типу спазмов длительностью по 3 секунды, возникающие сериями. Также нередко приступы начинались с икоты. После назначения фенобарбитала и депакино хроно было отмечено урежение частоты приступов, однако ремиссия достигнута не была. В настоящее время у пациентки возникает 2 типа эпилептических приступов: первый тип – заведение глаз с вертикальным инстагмом и легким запрокидыванием головы (до 5 р/сут), второй тип – заведением глаз с тоническим напряжением туловища и конечностей: сгибание ручек в локтевых суставах и заведением их за голову, иногда с приведением к животу ножек, обычно после пробуждения [4, с.63].
Из анамнеза жизни известно, что ребенок от второй беременности (при первой – выкидыш). Беременность постоянно протекала с угрозой прерывания. Роды на 36 неделе гестации. Девочка родилась с весом 2,5 кг, окружностью головы 33 см. ребенок развивался с выраженной задержкой психического и моторного развития. При этом семейный анамнез не отягощен.
При осмотре определяется выраженный краниофациальный дисморфизм: микроцефалия, выступающий лоб, впалые виски, низко посаженные оттопыренные ушные раковины, короткий нос с широкой переносицей и вздернутым кончиком, эпикант, маленькая нижняя челюсть, укорочение дистальных фаланг пальцев рук.
Неврологический статус: практическое полное отсутствие развития ребенка: с трудом контролирует голову, не ползает, не может переворачиваться, не сидит. Взгляд прослеживает, периодически возникает «комплекс оживления» при виде матери; инструкции не выполняет, речь полностью отсутствует. Псевдобульбарный парез, глотает только жидкую пищу. Сходящееся косоглазие. Гипомимия. Тетрапарез, более выраженный в ногах, при диффузно сниженном мышечном тонусе. Сухожильные рефлексы равномерно повышены, зона расширена; определяются патологические стопные и кистевые пирамидные симптомы. Брюшные рефлексы угнетены. Тазовые функции не контролирует. В контакт не вступает, игрушками также не интересуется.
Видео-ЭЭГ мониторинг: биоэлектрическая активность головного мозга представлена высокоамплитудными диффузными медленными волнами в сочетании с медленными комплексами пик/полипик-волна, острая-медленная волна, низкой степени синхронизации. Также отмечается амплитудное преобладание эпилептиформной активности в задних отделах, максимально выраженное по правым затылочным электродам с реверсией фазы; амплитуда уменьшается по мере приближения к лобным отделам, где эпилептиформные проявления минимальны. В состоянии бодрствования зарегистрированы тонические эпилептические спазмы, исходящие из правых затылочных отведений.
При проведении МРТ была выявлена тотальная агирия-лиссэнцефалия. Кортикальная складчатость резко обеднена, основные борозды и извилины не сформированы. Кора патологически утолщена; в субкортикальном белом веществе регистрируется диспластическая дисмиелинизация, объем белого вещества снижен. Боковые желудочки диспластически расширены [4, с.64].
Заключение
Таким образом, в ходе работы ознакомилась с проявлением синдрома Миллера-Дикера на рассмотренном клиническом случае, который характеризуется краниофациальными аномалиями, выраженной психической и моторной задержкой в развитии, а также ранним дебютом эпилептических приступов. Синдром может быть установлен при сочетании с классической лиссэнцефалией. Также большинство детей умирает до раненого детского возраста. Поэтому родителям необходимо проходить такие исследования, как МРТ, картирование.
Список литературы:
1. Бобылова М.Ю., Миронов М.Б, Абрамов, М.О, Куликов А.В, Казакова М.В, Глухова Л.Ю, Барлетова Е.И, Мухин К.Ю. Клинический случай мутации гена SYNGAP1 у девочки с эпилепсией, умственной отсталость, аутизмом и двигательными нарушениями//Русский журнал детской неврологии. – 2015. — Т 10. — №3. — с. 49.
2. Заведенко Н.Н, Мутовин Г.Р. Эпилепсия при наследственной патологии//Эпилепсия и пароксизмальные состояния. – 2013. — Т 5. — №4. — с. 35.
3. Маланина Е.Н, Касымова Д.Р, Азизов Р.Р, Соколова Ю.В. Пренатальная диагностика редкого синдрома с широким спектром мальформаций головного мозга: описание случая, дифференциальная диагностика//Пренатальная диагностика. – 2011.- Т 10. — №2. — с. 142.
4. Мухин К.Ю, Безрукова И.С, Тягун Н.С, Миронов М.Б, Алиханов А.А, Петрухин А.С. Синдром Миллера-Дикера//Русский журнал детской неврологии. – 2008. — Т 3. — №3. — с. 61-66.
5. Тысячина М.Д, Мухин К.Ю, Какаулина В.С, Кузина Н.Ю, Морозов Д.В, Барлетова Е.И, Миронов М.Б, Петрухин А.С. Эпилептические приступы при синдроме трисомии Х (описание случая)//Русский журнал детской неврологии. – 2009. — Т 4. — №4. — с. 38.
Источник