У меня синдром романо уорда

Синдром удлиненного QT (LQT): причины, диагностика, лечениеЭтиология и встречаемость синдрома удлиненного QT. Синдромы удлиненного QT (LQT) — разнородная панэтническая группа нарушений, получивших название каналопатий, поскольку они вызываются дефектами в ионных каналах сердца. Распространеность синдромов удлиненного QT приблизительно 1 на 5000-7000 человек. Большинство случаев удлиненного QT вызвано мутациями в пяти известных генах ионных каналов сердца (KCNQ1, KCNH2, SCN5A, KCNE1.KKCNE2). Генетика, лежащая в основе синдромов удлиненного QT, сложна. Во-первых, существует локусная гетерогенность. Наиболее частый из синдромов удлиненного QT, аутосомно-доминантный синдром Романо-Уорда (MIM №192500), вызван преимущественно мутациями в двух локусах, KCNQ1 и KCNH2, а также содействующим третьим локусом, SCN5A. Во-вторых, разные мутантные аллели в одном и том же локусе могут вызывать два различающихся синдрома удлиненного QT, синдром Романо-Уорда и аутосомно-рецессивный синдром Джервелла-Ланге-Нильсена (MIM №220400). Патогенез синдрома удлиненного QTСиндром удлиненного QT вызывается дефектами реполяризации в клетках сердца. Реполяризация — управляемый процесс, требующий баланса между направленным внутрь клетки потоком натрия и кальция и из клетки — калия. Дисбаланс удлиняет или укорачивает длительность потенциала действия, вызывающего соответственно удлинение или сокращение интервала QT на электрокардиограмме. Большинство случаев синдрома удлиненного QT вызваны мутациями с утратой функции в генах, кодирующих субъединицы или полные белки каналов калия (названия этих генов начинаются с KCN). Эти мутации уменьшают реполяризацию, тем самым продлевая потенциал действия клетки и уменьшая порог для последующей деполяризации. У других пациентов с синдромом удлиненного QT мутации с усилением функции в гене натриевого канала, SCN5A, ведут к повышенному притоку натрия, вызывая аналогичные изменения потенциала действия и эффекты реполяризации.
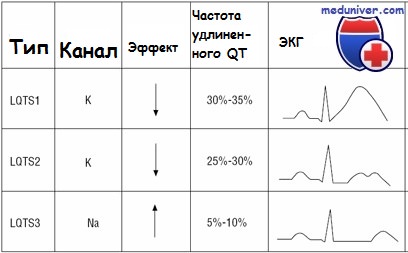
Фенотип и развитие синдрома удлиненного QTСиндромы удлиненного QT характеризуются удлинением интервала QT и аномалиями зубца Т на электрокардиограмме, включая тахиаритмию и полиморфную желудочковую тахикардию. Желудочковая тахикардия характеризуется изменением амплитуды и скручиванием комплекса QRS. Полиморфная желудочковая тахикардия связана с удлиненным интервалом QT и обычно заканчивается спонтанно, но может упорствовать и прогрессировать в фибрилляцию желудочков. При самом частом варианте синдрома удлиненного QT, Романо-Уорда, обмороки из-за аритмии сердца — наиболее частый признак. Если ребенок остается недиагностированным или не получает лечение, синкопальные состояния повторяются и могут быть летальными в 10-15% случаев. Тем не менее от 30 до 50% индивидуумов с синдромом удлиненного QT никогда не имеют синкопальных симптомов. Сердечные эпизоды чаще всего встречаются в возрасте от 9 до 12 лет, уменьшаясь со временем. Эпизоды могут происходить в любом возрасте, если спровоцированы приемом медикаментов, удлиняющих интервал QT. Нефармакологические триггеры сердечных событий при синдроме Романо-Уорда отличаются в зависимости от ответственного гена. Триггеры LQT1 — обычно адренергические стимулы, включая физическую нагрузку и внезапные эмоции (испуг). Лица с LTQ2 находятся в риске как при нагрузке, так и в покое, а также при слуховых стимулах, например звонок будильника или телефона. Пациенты с LQT3 имеют эпизоды с замедлением сердечных показателей в периоды отдыха и сна. Кроме того, 40% случаев LQT1 проявляют себя до 10-летнего возраста; симптоматика появляется до 10 лет жизни только в 10% случаев LTQ2 и крайне редко при LQT3. Синдром LQT5 редкий, о течении и триггерах известно меньше. Синдром удлиненного QT имеет неполную пенетрантность, как с точки зрения электрокардиографических аномалий, так и синкопальных эпизодов. До 30% больных могут иметь интервалы QT, перекрывающиеся с нормальными колебаниями. Варьирующаяся экспрессия заболевания может происходить как внутри семьи, так и между семьями. Из-за неполной пенетрантности для точного диагноза у членов семьи часто необходима нагрузочная электрокардиография. Синдромы удлиненного QT могут сопровождаться дополнительными данными при медицинском осмотре. Например, синдром Джервелла-Ланге-Нильсена (MIM №220400) характеризуется глубокой врожденной нейросенсорной глухотой в сочетании с синдромом удлиненного QT. Это аутосомно-рецессивное заболевание, вызываемое также определенными мутациями в одном из двух генов (KCNQ1 и KCNE1), участвующих в развитии аутосомно-доминантного синдрома Романо-Уорда. Гетерозиготные родственники пациентов с синдромом Джервелла-Ланге-Нильсена не глухие, но имеют 25% риск развития синдрома удлиненного QT. Особенности фенотипических проявлений синдрома удлиненного QT:
Лечение синдрома удлиненного QTЛечение синдрома удлиненного QT направлено на предотвращение синкопальных эпизодов и остановки сердца. Оптимальное лечение зависит от идентификации ответственного в данном случае гена. Например, терапия b-адреноблокаторами до начала симптомов — наиболее эффективный метод при LQT1 и, отчасти, при LQT2, но его эффективность при LQT3 незначительна. При лечении b-адреноблокаторами необходимо тщательно проверять соответствие возрастным дозам, не прерывать прием лекарственных средств. Для больных с брадикардией могут оказаться необходимыми водители ритма; может потребоваться доступ к внешним дефибрилляторам. Имплантируемые кардиовертеры-дефибрилляторы могут быть необходимыми больным с LQT3 или некоторым лицам с синдромом удлиненного QT, для которых проблематична терапия бета-адреноблокаторами, например больным бронхиальной астмой, депрессией или сахарным диабетом, а также пациентам с остановкой сердца в анамнезе. Некоторые лекарства, например антидепрессивный препарат амитриптилин, фенилэфрин и дифенилгидрамин, или противогрибковые лекарства, включая флуконазол и кетоназол, должны быть исключены из-за их действия, удлиняющего интервал QT или повышения симпатико-тонии. Исключают также виды деятельности и спорта, связанные с интенсивной физической нагрузкой и эмоциональным стрессом.
Риски наследования синдрома удлиненного QTЛица с синдромом Романо-Уорда имеют 50% шанс родить ребенка с унаследованными мутациями в гене. Поскольку частота новых мутаций низкая, большинство больных имеют пораженного родителя (хотя, возможно, бессимптомного). Чрезвычайно важны и могут оказаться жизнесохраняющими подробный семейный анамнез и тщательная кардиологическая оценка членов семьи. Риск повторения для сибсов пациентов с синдромом Джервелла-Ланге-Нильсена — 25%, как и ожидается при аутосомно-рецессивном заболевании. Пенетрантность изолированного синдрома удлиненного QT без глухоты для гетерозиготных носителей синдрома Джервелла-Ланге-Нильсена — 25%. Пример синдрома удлиненного QT. А.Б., 30-летняя женщина с синдромом удлиненного QT (LQT), обратилась в генетическую клинику вместе с мужем, поскольку они планируют беременность. Пара хочет знать риск повторения этого заболевания у детей и подходящие методы генетического тестирования и пренатальной диагностики. Женщина также обеспокоена потенциальным влиянием беременности на ее собственное здоровье. Диагноз синдрома LQT установлен в начале третьего десятилетия жизни, когда она проходила обследование после внезапной смерти ее 15-летнего брата. В целом она — здоровый человек с нормальным слухом, отсутствием дисморфических признаков. У нее никогда не было обморочных состояний. Впоследствии электрокардиографические данные подтвердили диагноз синдрома у нее, ее отца и одной из теток по отцу. Молекулярное тестирование выявило миссенс-мутацию в гене KCNH2, ранее описанную в других семьях с синдромом Романо-Уорда, тип LQT2. Первоначально пациентка получала бета-адреноблокаторы, но ее кардиологи решили, что низкая эффективность b-адреноблокаторов при LQT2 и летальный случай у ее брата оправдывают имплантацию кардиовертера-дефибриллятора как ей, так и ее пораженным родственникам. Пациентка — первый человек в ее семье, проходящий генетическое консультирование по синдрому удлиненнго QT. — Также рекомендуем «Синдром Марфана: причины, диагностика, лечение» Оглавление темы «Наследственные болезни»:
|
Источник
| синдром Романо-Уорда | |
|---|---|
| Схематическое изображение нормальной кривой ЭКГ ( синусового ритма ), с волнами, сегменты и интервалы мечеными. | |
| причины | Мутации в Kcnq1, KCNH2, и гены SCN5A |
| диагностический метод | ЭКГ, нагрузочный тест |
| лечение | Бета-адренергические блокада |
Синдром Романо-Уорда ( RWS ) является наиболее распространенным подтипом синдрома длительного интервала QT , наследственным заболеванием сердца , которое влияет на электрические свойства клеток сердечной мышцы. Условие делает тех , кто пострадал в опасности аномальных сердечных ритмов , которые могут привести к обморокам , судороги , или внезапной смерти .
Синдром Романо-Уорда можно отличить клинически от других форм длительного интервала QT, воздействуя только на электрические свойства сердца, в то время как другие формы длительного интервала QT могут также влиять на другие части тела. Условие можно лечить с помощью лекарств , таких как бета-блокаторы , с ИКД , или хирургического вмешательства , чтобы сорвать симпатическую нервную систему .
Признаки и симптомы
синдром Романо-Уорда представляет следующее пораженного человека:
- Мерцание желудочков
- синкопа
- Torsade де пуантах
- Ненормальность уха
генетика
Синдром Романо-Уорд является описательным термином для группы подтипов длительного интервала QT синдрома, в частности , подтипы LQT1-6 и LQT9-16. Эти подтипы характеризуются аутосомно — доминантным наследованием и только влияют на электрическую активность сердца. Другие формы длительного интервала QT , включают синдром Джервелл-Ланге-Нильсена , характеризующийся аутосомно — рецессивным наследованием и врожденной глухотой , синдром Андерсона-Тавил (LQT7) , который сочетает в себе длительный QT интервал с периодическим параличом и ненормально формы лица, и синдрома Тимоти (LQT8) которая сочетает в себе длительный интервал QT, аномалии в строении сердца и расстройства аутистического спектра .
Таблица причинных генов
| Тип | OMIM | Ген | Заметки |
| LQT1 | 192500 | KCNQ1 | Кодирует альфа-субъединицу медленного задержанного канала выпрямителя калия К V 7.1 , несущей калий ток I Ks . |
| LQT2 | 152427 | KCNH2 | Также известен как HERG. Кодирует альфа-субъединицу быстрого задержанного канала выпрямителя калия К V 11.1 , несущего калий тока I Kr . |
| LQT3 | 603830 | SCN5A | Кодирует е альфа-субъединицу сердечного натриевого канала Na V 1,5 , несущего натрий ток I Na . |
| LQT4 | 600919 | ank2 | Зашифровывает анкириновые B, который закрепляет ионные каналы в клетке. Спорные истинная связь с QT пролонгации. |
| LQT5 | 176261 | KCNE1 | Зашифровывает норка, калиевые каналы β-субъединица. |
| LQT6 | 603796 | KCNE2 | Зашифровывает MiRP1, калиевые каналы β-субъединица. |
| LQT9 | 611818 | CAV3 | Зашифровывает Кавеолины-3 отвечает за формирование мембранных мешочков , известные как кавеол. Мутации в этом гене могут увеличить поздний натрий я Na . |
| LQT10 | 611819 | SCN4B | Кодирует β4-субъединицу сердечного натриевого канала. |
| LQT11 | 611820 | AKAP9 | Закодировать-киназы ассоциированный белок , который взаимодействует с K V 7.1. |
| LQT12 | 601017 | SNTA1 | Зашифровывает syntrophin-& alpha ; 1. Мутации в этом гене могут увеличить поздний ток натрия я Na . |
| LQT13 | 600734 | KCNJ5 | Также известен как GIRK4 , кодирует белок G-чувствительный внутрь выпрямляющие калиевые каналы (К л 3.4) , которые несут калий тока I K (АХ) . |
| LQT14 | 616247 | CALM1 | Зашифровывает кальмодулин-1, кальций-связывающий белок мессенджер , который взаимодействует с током кальция я Ca (L) . |
| LQT15 | 616249 | CALM2 | Зашифровывает кальмодулин-2, кальций-связывающий белок мессенджер , который взаимодействует с током кальция я Ca (L) . |
| LQT16 | 114183 | CALM3 | Зашифровывает кальмодулин-3, кальций-связывающий белок мессенджер , который взаимодействует с током кальция я Ca (L) . |
Механизм
В формах Романо-Уорда синдрома длительного интервала QT, генетические мутации затрагивают , как положительно заряженные ионы , такие как ионы калия, натрия и кальция транспортируются в и из клеток сердца . Многие из этих генов кодируют белки , которые образуют или взаимодействуют с ионными каналами . В сердечной мышце, эти ионные каналы играют важную роль в поддержании нормального ритма сердца. Мутации в любом из этих генов изменяют структуру или функцию каналов, которая изменяет поток ионов между клетками, срыв в транспорте ионов изменяет способ сердечных сокращений, что приводит к ненормальному сердечному ритму , характерным для синдрома.
Белок, вырабатываемый ank2 гена гарантирует , что другие белки, в частности , ионные каналы, которые вставлены в клеточную мембрану соответственно. Мутация в гене ank2 вероятно , изменяет поток ионов между клетками в сердце, что нарушает нормальный ритм сердца и приводит к особенностям синдрома Романо-Уорда.
диагностика
С точки зрения диагностики синдрома Романо-Уорда следующее сделано , чтобы установить состояние (The Score Schwartz помогает в этом):
- Упражнение тест
- ЭКГ
- История семьи
лечение
Лечение синдрома Романо-Уорда может справиться с дисбалансом между правой и левой стороны симпатической нервной системы , которые могут играть определенную роль в деле этого синдрома. Дисбаланс может быть временно отменено с левым блоком звездчатого ганглия, который укоротить интервал QT. Если это успешно, хирургическая ганглиэктомия может быть выполнена в виде постоянной аритмии treatment.Ventricular может управляться бета-адренергических блокадами ( пропранолол )
эпидемиология
синдром Романо-Уорда является наиболее распространенной формой наследственного длительного интервала QT синдрома, затрагивающей около 1 в 7000 человек по всему миру.
Смотрите также
- Длинные QT синдром
- Джервелл и Ланге-Нильсена синдром
- Синдром Андерсена-Тавил
- синдром Тимоти
Рекомендации
дальнейшее чтение
- Келли, Эвелин Б. (2013-01-07). Энциклопедия генетики человека и болезней . ABC-CLIO. ISBN 9780313387135 . извлекаться 2017-04-07
- Накано, Юкико; Shimizu, Wataru (2016-01-01). «Генетика долгосрочной QT синдрома» . Журнал генетики человека . 61 (1): 51-55. DOI : 10.1038 / jhg.2015.74 . ISSN 1434-5161 . PMID 26108145 .
внешняя ссылка
Источник
4-я хромосо́ма челове́ка — одна из 23 человеческих хромосом. Хромосома содержит более 191 млн пар оснований[1], что составляет примерно 6—6,5 % всего материала ДНК человеческой клетки. В настоящее время считается, что на 4-й хромосоме находятся от 700 до 1100 генов.
Гены[править | править код]
Ниже перечислены некоторые гены, расположенные на 4-й хромосоме:
- CXCL1 — CXC-хемокин 1;
- CXCL2 — CXC-хемокин 2;
- CXCL3 — CXC-хемокин 3;
- CXCL4 — CXC-хемокин 4 (фактор тромбоцитов 4);
- CXCL5 — CXC-хемокин 5;
- CXCL6 — CXC-хемокин 6 (гранулоцитарный хемотактический белок 2);
- CXCL7 — CXC-хемокин 7;
- CXCL9 — CXC-хемокин 9;
- CXCL10 — CXC-хемокин 10;
- CXCL11 — CXC-хемокин 11;
- CXCL13 — CXC-хемокин 13;
- EVC — белок, ассоциированный с синдромом Эллиса — ван Кревельда;
- EVC2 — белок, ассоциированный с синдромом Эллиса — ван Кревельда 2;
- FGFR3 — рецептор фактора роста фибробластов 3;
- FGFRL1 — рецептор, подобный рецептору фактора роста фибробластов 1;
- CFI — фактор комплемента I;
- HTT — хантингтин;
- MMAA — белок, ассоциированный с метилмалоновой ацидурией;
- PHOX2B — гомеодомен фактора транскрипции;
- PKD2 — белок, ассоциированный с аутосомно-доминантной поликистозной болезнью почек 2;
- PLK4;
- QDPR — хиноидная дигидроптеридин-редуктаза;
- SNCA — α-синуклеин;
- UCHL1 — убиквитин-карбокси-концевая гидролаза L1 (убиквитин тиолэстераза);
- WFS1 — вольфрамин;
- FGF2 — фактор роста фибробластов 2 (основной фактор роста фибробластов);
- KDR — киназный инсерционный домен-содержащий рецептор (рецептор васкулярного эндотелиального фактора роста 2);
- IGJ — белок, соединяющий α- и μ-субъединицы иммуноглобулинов (J-цепь иммуноглобулинов).
Плечо p[править | править код]
- CRMP1 — медиатор коллапсина 1, член семейства медиаторов CRMP (англ. CRMP family);
- SOD3 — Супероксиддисмутаза 3.
Плечо q[править | править код]
- ANK2 — адаптерный белок семейства анкиринов, анкирин B;
- CLCN3 — Хлоридный канал 3;
- ENPEP — глутамиламинопептидаза (CD249);
- CLTB — клатрин B;
- CXCL8 — CXC-хемокин 8 (интерлейкин 8);
- IL2 — интерлейкин 2;
- IL8 — интерлейкин 8.
Болезни и расстройства[править | править код]
Ниже перечислены некоторые заболевания связанные с генами 4-й хромосомы, а также гены, дефекты которых вызывают эти заболевания:
- поликистоз почек — PKD2;
- ахондроплазия — FGFR3;
- бессиндромная глухота (англ. nonsyndromic deafness), аутосомно-доминантный тип 6 — WFS1;
- болезнь Гиршпрунга — PHOX2B;
- болезнь Паркинсона — SNCA;
- болезнь Хантингтона — HTT;
- гемолитико-уремический синдром (англ. hemolytic-uremic syndrome) — CFI;
- гемофилия C — F11;
- гипохондроплазия — FGFR3;
- дефицит тетрагидробиоптерина (англ. tetrahydrobiopterin deficiency) — QDPR;
- метилмалоновая ацидемия (англ. methylmalonic acidemia), тип cblA — MMAA;
- рак мочевого пузыря — FGFR3;
- себоррейный кератоз (англ. seborrheic keratosis) — FGFR3;
- синдром Вольфрама, тип 1 — WFS1;
- синдром длинного интервала QT, тип 4 — ANK2;
- синдром Крузона с чёрным акантозом (англ. Crouzonodermoskeletal syndrome) — FGFR3;
- синдром Муэнке (англ. Muenke syndrome) — FGFR3;
- синдром проклятия Ундины — PHOX2B;
- синдром Романо — Уорда (англ. Romano–Ward syndrome) — ANK2;
- синдром Эллиса — ван Кревельда (англ. Ellis–van Creveld syndrome) — EVC и EVC2;
- танатофорная карликовость (англ. thanatophoric dysplasia), типы 1 и 2 — FGFR3;
- тяжёлая ахондроплазия с задержкой развития и чёрным акантозом (англ. severe achondroplasia with developmental delay and acanthosis nigricans [SADDAN]) — FGFR3;
- хронический лимфолейкоз.
Примечания[править | править код]
- ↑
Human chromosome 4 map view (англ.). Vertebrate Genome Annotation (VEGA) database. The Wellcome Trust Sanger Institute. — Карта хромосомы и её основные параметры: размер, количество генов и т. п. Дата обращения 18 августа 2009. Архивировано 6 апреля 2012 года.
Источник
| Romano–Ward syndrome | |
|---|---|
| Schematic representation of normal ECG trace (sinus rhythm), with waves, segments, and intervals labeled. | |
| Symptoms | Faint, seizure[1] |
| Causes | Mutations in the KCNQ1, KCNH2, and SCN5A genes [2] |
| Diagnostic method | EKG, Exercise test[3] |
| Treatment | Beta-adrenergic blockade [4] |
Romano–Ward syndrome is the most common form of congenital Long QT syndrome (LQTS), a genetic heart condition that affects the electrical properties of heart muscle cells. [5] Those affected are at risk of abnormal heart rhythms which can lead to fainting, seizures, or sudden death.[6][2][7] Romano–Ward syndrome can be distinguished clinically from other forms of inherited LQTS as it affects only the electrical properties of the heart, while other forms of LQTS can also affect other parts of the body.
Romano–Ward syndrome is caused by abnormal variants in the genes responsible for producing certain proteins used to transport charged particles (ion channels) within the heart.[5] These abnormalities interfere with the electrical signals that heart cells use to coordinate contractions, causing the heart to take longer to recharge in between beats. The condition is usually diagnosed using an electrocardiogram, but other tests sometimes used include Holter monitoring, exercise testing, and genetic testing.[1] It may be treated using medication such as beta-blockers, an implantable cardioverter-defibrillator, or surgery to disrupt the sympathetic nervous system.[8] Romano–Ward syndrome is estimated to affect 1 in every 7,000 people.
Signs and symptoms[edit]
Romano–Ward syndrome increases the risk of abnormal heart rhythms or arrhythmias. These are typically a form of ventricular tachycardia known as Torsades de Pointes which can cause faints, seizures, or even sudden death.[1] Less dangerous arrhythmias such as atrial fibrillation also occur, causing symptoms of heart racing or palpitations. However, many of those with Romano–Ward syndrome will remain free from arrhythmias and therefore free from symptoms. Certain situations are more likely to precipitate arrhythmias such as exercise or mental stress in the LQT1 subtype, sudden loud noise in the LQT2 subtype, and during sleep or immediately upon waking in the LQT3 subtype.[9]
Romano–Ward syndrome can be differentiated from other forms of long QT syndrome by Romano-Ward’s sole involvement of the heart. While other forms of long QT syndrome are associated with deafness (Jervell and Lange-Nielsen syndrome), intermittent weakness and bone abormailities (LQT7, Andersen-Tawil syndrome), and autism spectrum disorder (LQT8, Timothy syndrome), these extra-cardiac manifestations are not seen in Romano-Ward.[8]
Causes[edit]
Romano–Ward syndrome is a descriptive term for a group of subtypes of long QT syndrome, specifically subtypes LQT1-6 and LQT9-16.[8] Several subtypes of Romano–Ward syndrome have been described based on the underlying genetic variant.[5] These subtypes differ in clinical presentation and their response to treatment. There is robust evidence that the genetic variants associated with the three most common subtypes (LQT1, LQT2 and LQT3) are truly causative of the syndrome. However, there is uncertainty as to whether some of the other rarer subtypes are truly disease-causing by themselves or instead make individuals more susceptible to QT prolongation in response to other factors such as medication or low blood potassium levels (hypokalaemia).[10]
LQT1[edit]
LQT1 is the most common subtype of Romano–Ward syndrome, responsible for 30 to 35% of all cases.[5] The gene responsible, KCNQ1, has been isolated to chromosome 11p15.5 and encodes the alpha subunit of the KvLQT1 potassium channel. This subunit interacts with other proteins (in particular, the minK beta subunit) to create the channel, which carries the delayed potassium rectifier current IKs responsible for the repolarisation phase of the cardiac action potential.[5]
Variants in KCNQ1 cause the LQT1 subtype of Romano–Ward syndrome when a single copy of the variant is inherited (heterozygous, autosomal dominant inheritance). When two copies of the variant are inherited (homozygous, autosomal recessive inheritance) the more severe Jervell and Lange-Nielsen syndrome is found, associated with more marked QT prolongation, congenital sensorineural deafness, and a greater risk of arrhythmias.[5]
LQT1 is associated with a high risk of faints but lower risk of sudden death than LQT2.[citation needed]
LQT1 may also affect glucose regulation. After ingesting glucose, those with LQT1 produce more insulin than would be expected, which is followed by a period of insulin resistance. When the resistance diminishes, abnormally low blood glucose levels (hypoglycaemia) are sometimes seen.[11]
LQT2[edit]
The LQT2 subtype is the second-most common form of Romano–Ward syndrome, responsible for 25 to 30% of all cases.[5] This form of Romano–Ward syndrome is caused by variants in the KCNH2 gene on chromosome 7.[5]KCNH2 (also known as hERG) encodes the potassium channel which carries the rapid inward rectifier current IKr. This current contributes to the terminal repolarisation phase of the cardiac action potential, and therefore the length of the QT interval.[5]
LQT3[edit]
The LQT3 subtype of Romano–Ward syndrome is caused by variants in the SCN5A gene located on chromosome 3p21-24. SCN5A encodes the alpha subunit of the cardiac sodium channel, NaV1.5, responsible for the sodium current INa which depolarises cardiac cells at the start of the action potential.[5] Cardiac sodium channels normally inactivate rapidly, but the mutations involved in LQT3 slow their inactivation leading to a small sustained ‘late’ sodium current. This continued inward current prolongs the action potential and thereby the QT interval.[5]
A large number of mutations have been characterized as leading to or predisposing to LQT3. Calcium has been suggested as a regulator of SCN5A protein, and the effects of calcium on SCN5A may begin to explain the mechanism by which some of these mutations cause LQT3. Furthermore, mutations in SCN5A can cause Brugada syndrome, cardiac conduction disease, and dilated cardiomyopathy. In rare situations, some affected individuals can have combinations of these diseases.[citation needed]
Other subtypes[edit]
LQT5 is caused by variants in the KCNE1 gene. This gene is responsible for the potassium channel beta subunit MinK which, in conjunction with the alpha subunit encoded by KCNQ1, is responsible for the potassium current IKs, and variants associated with prolonged QT intervals decrease this current.[5] The same variants in KCNE1 can cause the more severe Jervell and Lange-Nielsen syndrome when two copies are inherited (homozygous inheritance) and the milder LQT5 subtype of Romano–Ward syndrome when a single copy of the variant is inherited (heterozygous inheritance).[12]
The LQT6 subtype is caused by variants in the KCNE2 gene.[5] This gene is responsible for the potassium channel beta subunit MiRP1 which generates the potassium current IKr, and variant that decrease this current have been associated with prolongation of the QT interval.[12] However, subsequent evidence such as the relatively common finding of variants in the gene in those without long QT syndrome, and the general need for a second stressor such as hypokalaemia to be present to reveal the QT prolongation, has suggested that this gene instead represents a modifier to susceptibility to QT prolongation.[13] Some, therefore, dispute whether variants in the gene are sufficient to cause Romano–Ward syndrome by themselves.[13]
LQT9 is caused by variants in the membrane structural protein, caveolin-3.[5] Caveolins form specific membrane domains called caveolae in which voltage-gated sodium channels sit. Similar to LQT3, these caveolin variants increase the late sustained sodium current, which impairs cellular repolarization.[5]
LQT10 is an extremely rare subtype, caused by variants in the SCN4B gene. The product of this gene is an auxiliary beta-subunit (NaVβ4) forming cardiac sodium channels, variants in which increase the late sustained sodium current.[5] LQT13 is caused by variants in GIRK4, a protein involved in the parasympathetic modulation of the heart.[5] Clinically, the patients are characterized by only modest QT prolongation, but an increased propensity for atrial arrhythmias. LQT14, LQT15 and LQT16 are caused by variants in the genes responsible for calmodulin (CALM1, CALM2, and CALM3 respectively).[5] Calmodulin interacts with several ion channels and its roles include modulation of the L-type calcium current in response to calcium concentrations, and trafficking the proteins produced by KCNQ1 and thereby influencing potassium currents.[5] The precise mechanisms by which means these genetic variants prolong the QT interval remain uncertain.[5]
Table of causative genes
| Type | OMIM | Gene | Notes |
| LQT1 | 192500 | KCNQ1 | Encodes the α-subunit of the slow delayed rectifier potassium channel KV7.1 carrying the potassium current IKs.[10] |
| LQT2 | 152427 | KCNH2 | Also known as hERG. Encodes the α-subunit of the rapid delayed rectifier potassium channel KV11.1 carrying the potassium current IKr.[10] |
| LQT3 | 603830 | SCN5A | Encodes the e α-subunit of the cardiac sodium channel NaV1.5 carrying the sodium current INa.[10] |
| LQT4 | 600919 | ANK2 | Encodes Ankyrin B which anchors the ion channels in the cell. Disputed true association with QT prolongation.[10] |
| LQT5 | 176261 | KCNE1 | Encodes MinK, a potassium channel β-subunit.[10] |
| LQT6 | 603796 | KCNE2 | Encodes MiRP1, a potassium channel β-subunit.[10] |
| LQT9 | 611818 | CAV3 | Encodes Caveolin-3 responsible for forming membrane pouches known as caveolae. Mutations in this gene may increase the late sodium INa.[10] |
| LQT10 | 611819 | SCN4B | Encodes the β4-subunit of the cardiac sodium channel.[10] |
| LQT11 | 611820 | AKAP9 | Encode A-kinase associated protein which interacts with KV7.1.[10] |
| LQT12 | 601017 | SNTA1 | Encodes syntrophin-α1. Mutations in this gene may increase the late sodium current INa.[10] |
| LQT13 | 600734 | KCNJ5 | Also known as GIRK4, encodes G protein-sensitive inwardly rectifying potassium channels (Kir3.4) which carry the potassium current IK(ACh).[10] |
| LQT14 | 616247 | CALM1 | Encodes calmodulin-1, a calcium-binding messenger protein that interacts with the calcium current ICa(L).[10] |
| LQT15 | 616249 | CALM2 | Encodes calmodulin-2, a calcium-binding messenger protein that interacts with the calcium current ICa(L).[10] |
| LQT16 | 114183 | CALM3 | Encodes calmodulin-3, a calcium-binding messenger protein that interacts with the calcium current ICa(L).[10] |
Mechanism[edit]
In the Romano-Ward forms of Long QT syndrome, genetic mutations affect how positively-charged ions, such as potassium, sodium and calcium ions are transported in and out of heart cells. Many of these genes encode proteins which form or interact with ion channels. In cardiac muscle, these ion channels play critical roles in maintaining the heart’s normal rhythm. Mutations in any of these genes alter the structure or function of channels, which changes the flow of ions between cells, a disruption in ion transport alters the way the heart beats, leading to abnormal heart rhythm characteristic of the syndrome.[4][14][15][16]
The protein made by the ANK2 gene ensures that other proteins, particularly ion channels, are inserted into the cell membrane appropriately. A mutation in the ANK2 gene likely alters the flow of ions between cells in the heart, which disrupts the heart’s normal rhythm and results in the features of Romano–Ward syndrome.[medical citation needed]
Diagnosis[edit]
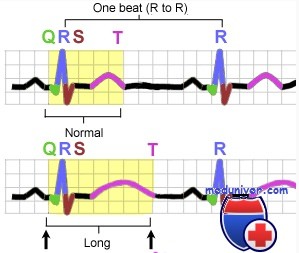
The normal range of QT intervals in the normal population and in those with Romano-Ward syndrome
Characteristic T-wave patterns in the 3 major subtypes of Romano-Ward syndrome
Romano–Ward syndrome is principally diagnosed by measuring the QT interval corrected for heart rate (QTc) on a 12-lead electrocardiogram (ECG). Romano–Ward syndrome is associated with a prolonged QTc, although in some genetically proven cases of Romano–Ward syndrome this prolongation can be hidden, known as concealed Long QT syndrome.[12] The QTc is less than 450 ms in 95% of normal males, and less than 460 ms in 95% of normal females. Romano–Ward syndrome is suggested if the QTc is longer than these cutoffs. However, as 5% of normal people also fall into this category, some suggest cutoffs of 470 and 480 ms for males and females respectively, corresponding with the 99th centiles of normal values.[12]
The major subtypes of Romano–Ward syndrome are associated with specific ECG features. LQT1 is typically associated with broad-based T-waves, whereas the T-waves in LQT2 are notched and of lower amplitude, whilst in LQT3 the T-waves are often late onset, being preceded by a long isoelectric segment.[12]
Other factors beyond the QT interval should be taken into account when making a diagnosis, some of which have been incorporated into scoring systems such as the Schwartz score.[3] These factors include a history of characteristic abnormal heart rhythms (Torsades de Pointes), unexplained blackouts (syncope), and a family history of confirmed LQT syndrome. Other investigations that may suggest a diagnosis of the LQT1 form of Romano–Ward syndrome include paradoxical lengthening of the QT interval in response to exercise (QTc >470 ms at 2–4 minutes of recovery) or during an artificial infusion of adrenaline (lengthening of the absolute QT interval >30 ms during low dose adrenaline).[12]
Treatment[edit]
The treatment for Romano–Ward syndrome aims to reduce the risk of arrhythmias. Lifestyle measures include avoiding very strenuous or competitive exercise.[1] Those with the LQT2 form of Romano–Ward syndrome should avoid sudden loud noises such as alarm clocks as these may trigger arrhythmias.[8] Fevers should be treated promptly with paracetamol.[1] Grapefruit juice should be avoided as it contains a chemical which decreases IKr and further prolongs the QT interval.[1]Medications that further prolong the QT interval such as sotalol should be avoided, lists of which can be found in publicly accessible online databases.[8]
Beta blockers such as propranolol or nadolol blunt the effects of adrenaline on the heart and thereby reduce the risk of arrhythmias.[8]Mexiletine, flecainide and ranolazine decrease the late sodium current and are of particular use in the LQT3 form of Romano–Ward syndrome,[8] and mexiletine may also be of benefit in other subtypes.[17] Potassium supplements may be used at times when potassium is being lost such as when experiencing diarrhoea or vomiting, but medications that encourage the retention of potassium such as spironolactone or amiloride may also be required.[1]
An implantable defibrillator, a small device that monitors the heart rhythm and can automatically deliver an electric shock to restart the heart, may be recommended. These devices are recommended for those with Romano–Ward syndrome who have experienced a cardiac arrest or a blackout whilst taking beta blockers.[8] In those who experience recurrent arrhythmias despite medical therapy, a surgical procedure called sympathetic denervation can be used to interrupt the nerves that stimulate the heart.[8]
Epidemiology[edit]
Romano–Ward syndrome is the most common form of inherited long QT syndrome, affecting an estimated 1 in 7,000 people worldwide.[citation needed]
See also[edit]
- Long QT syndrome
- Jervell and Lange-Nielsen syndrome
- Andersen-Tawil syndrome
- Timothy syndrome
References[edit]
- ^ a b c d e f g Tester DJ, Schwartz PJ, Ackerman MJ (2013). Gussak I, Antzelevitch C (eds.). Congenital Long QT Syndrome. Electrical Diseases of the Heart: Volume 1: Basic Foundations and Primary Electrical Diseases. Springer London. pp. 439–468. doi:10.1007/978-1-4471-4881-4_27. ISBN 9781447148814.
- ^ a b Reference, Genetics Home. «Romano-Ward syndrome». Genetics Home Reference. Retrieved 2017-04-01.
- ^ a b Mizusawa, Yuka; Horie, Minoru; Wilde, Arthur AM (2014-01-01). «Genetic and Clinical Advances in Congenital Long QT Syndrome». Circulation Journal. 78 (12): 2827–2833. doi:10.1253/circj.CJ-14-0905. PMID 25274057.
- ^ a b RESERVED, INSERM US14 — ALL RIGHTS. «Orphanet: Romano Ward syndrome». www.orpha.net. Retrieved 2017-04-01.
- ^ a b c d e f g h i j k l m n o p q r s t Bohnen MS, Peng G, Robey SH, Terrenoire C, Iyer V, Sampson KJ, Kass RS (January 2017). «Molecular Pathophysiology of Congenital Long QT Syndrome». Physiol. Rev. 97 (1): 89–134. doi:10.1152/physrev.00008.2016. PMC 5539372. PMID 27807201.
- ^ «Long QT syndrome 1 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program». rarediseases.info.nih.gov. Retrieved 2018-04-17.
- ^ Alders, Mariëlle; Christiaans, Imke (1993-01-01). «Long QT Syndrome». In Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Ledbetter, Nikki; Mefford, Heather C. (eds.). GeneReviews. Seattle (WA): University of Washington, Seattle. PMID 20301308.update 2015
- ^ a b c d e f g h i Priori, S. G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P. M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; Kirchhof, P.; Kjeldsen, K.; Kuck, K. H.; Hernandez-Madrid, A.; Nikolaou, N.; Norekvål, T. M.; Spaulding, C.; Van Veldhuisen, D. J.; Task Force for the Management of Patients with Ventricular Arrhythmias the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) (29 August 2015). «2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death». Europace. 17 (11): 1601–87. doi:10.1093/europace/euv319. ISSN 1099-5129. PMID 26318695.
- ^ Nakajima T, Kaneko Y, Kurabayashi M (2015). «Unveiling specific triggers and precipitating factors for fatal cardiac events in inherited arrhythmia syndromes». Circulation Journal. 79 (6): 1185–92. doi:10.1253/circj.CJ-15-0322. PMID 25925977.
- ^ a b c d e f g h i j k l m n o Giudicessi, John R.; Wilde, Arthur A. M.; Ackerman, Michael J. (October 2018). «The genetic architecture of long QT syndrome: A critical reappraisal». Trends in Cardiovascular Medicine. 28 (7): 453–464. doi:10.1016/j.tcm.2018.03.003. ISSN 1873-2615. PMC 6590899. PMID 29661707.
- ^ Demirbilek H, Galcheva S, Vuralli D, Al-Khawaga S, Hussain K (May 2019). «Ion Transporters, Channelopathies, and Glucose Disorders». Int J Mol Sci. 20 (10): 2590. doi:10.3390/ijms20102590. PMC 6566632. PMID 31137773.
- ^ a b c d e f Giudicessi JR, Ackerman MJ (2013). «Genotype- and Phenotype-Guided Management of Congenital Long QT Syndrome». Curr Probl Cardiol. 38 (10): 417–455. doi:10.1016/j.cpcardiol.2013.08.001. PMC 3940076. PMID 24093767.
- ^ a b Giudicessi JR, Wilde AA, Ackerman MJ (October 2018). «The genetic architecture of long QT syndrome: A critical reappraisal». Trends in Cardiovascular Medicine. 28 (7): 453–464. doi:10.1016/j.tcm.2018.03.003. PMC 6590899. PMID 29661707.
- ^ «ANK2 ankyrin 2 [Homo sapiens (human)] — Gene — NCBI». www.ncbi.nlm.nih.gov. Retrieved 2017-04-06.
- ^ «KCNE1 potassium voltage-gated channel subfamily E regulatory subunit 1 [Homo sapiens (human)] — Gene — NCBI». www.ncbi.nlm.nih.gov. Retrieved 2017-04-06.
- ^ «KCNE2 potassium voltage-gated channel subfamily E regulatory subunit 2 [Homo sapiens (human)] — Gene — NCBI». www.ncbi.nlm.nih.gov. Retrieved 2017-04-06.
- ^ G, Li; L, Zhang (November 2018). «The Role of Mexiletine in the Management of Long QT Syndrome». Journal of electrocardiology. PMID 30497731. Retrieved 2020-06-01.
Further reading[edit]
- Kelly, Evelyn B. (2013-01-07). Encyclopedia of Human Genetics and Disease. ABC-CLIO. ISBN 9780313387135. retrieved 2017-04-07
- Nakano, Yukiko; Shimizu, Wataru (2016-01-01). «Genetics of long-QT syndrome». Journal of Human Genetics. 61 (1): 51–55. doi:10.1038/jhg.2015.74. ISSN 1434-5161. PMID 26108145.
External links[edit]
Источник