Скачать презентацию на тему синдромы

Презентация на тему: Хромосомные синдромы
Скачать эту презентацию
Скачать эту презентацию
№ слайда 1

Описание слайда:
Хромосомные синдромы
№ слайда 2
 называются комплексы множествен")
Описание слайда:
Хромосомными болезнями (хромосомными синдромами) называются комплексы множественных врожденных пороков развития, вызываемых числовыми (геномные мутации) или структурными (хромосомные мутации) изменениями хромосом, видимыми в световой микроскоп. Хромосомными болезнями (хромосомными синдромами) называются комплексы множественных врожденных пороков развития, вызываемых числовыми (геномные мутации) или структурными (хромосомные мутации) изменениями хромосом, видимыми в световой микроскоп.
№ слайда 3

№ слайда 4

Описание слайда:
Хромосомные аберрации и изменения количества хромосом, как и генные мутации, могут возникать на разных этапах развития организма. Если они возникают в гаметах родителей, то аномалия будет наблюдаться во всех клетках развивающегося организма (полный мутант). Хромосомные аберрации и изменения количества хромосом, как и генные мутации, могут возникать на разных этапах развития организма. Если они возникают в гаметах родителей, то аномалия будет наблюдаться во всех клетках развивающегося организма (полный мутант). Если аномалия возникает в процессе эмбрионального развития при дроблении зиготы, кариотип плода будет мозаичным. Мозаичные организмы могут содержать несколько (2, 3, 4 и более) клеточных клонов с различными кариотипами. Это явление может сопровождаться мозаицизмом во всех либо в отдельных органах и системах.
№ слайда 5

Описание слайда:
Этиологические факторы хромосомной патологии все виды хромосомных мутаций (хромосомные аберрации) и некоторые геномные мутации (изменения числа хромосом). У человека встречаются только 3 типа геномных мутаций: тетраплоидия, триплоидия и анеуплоидия. Из всех вариантов анеуплоидий у живорожденных встречаются только трисомии по аутосомам, полисомии по половым хромосомам (три-, тетра- и пентасомии), а из моносомий — только моносомия X-хромосомы.
№ слайда 6

Описание слайда:
Полиплоидия у человека На рисунке представлен триплоидный хромосомный набор человека. Триплоидия отмечается в 17% случаев спонтанных выкидышей и в 3% случаев мертворождений.
№ слайда 7

Описание слайда:
Примерно 170 из 1000 эмбрионов и плодов погибают до рождения, из них около 40% — вследствие влияния хромосомных нарушений. Примерно 170 из 1000 эмбрионов и плодов погибают до рождения, из них около 40% — вследствие влияния хромосомных нарушений. Тем не менее, значительная часть мутантов (носителей хромосомной аномалии) минует действие внутриутробного отбора. Хромосомные болезни у новорожденных детей встречаются с частотой примерно 2,4 случая на 1000 родившихся.
№ слайда 8

Описание слайда:
Патогенез хромосомных болезней Специфические эффекты связаны с изменением числа структурных генов, кодирующих синтез специфических белков (увеличение при трисомиях и уменьшение при моносомиях). Полуспецифические эффекты при хромосомных болезнях могут быть обусловлены изменением числа генов, представленных и в норме многочисленными копиями (гены тРНК, рРНК, гистоновых и рибосомных белков и т. п.). Неспецифические эффекты хромосомных аномалий связывают с содержанием гетерохроматина, играющего важную роль в делении клеток, их росте и других физиологических процессах.
№ слайда 9
№ слайда 10

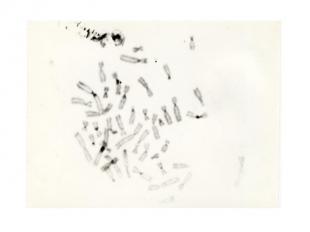
Описание слайда:
синдром Картагенера У детей с синдромом Картагенера (моногенное заболевание с аутосомно-рецессивным типом наследования, которое характеризуется триадой, включающей обратное расположение органов, бронхоэктазы и хронический синусит) были выявлены изменения околоцентромерных гетерохроматиновых районов хромосом (С-гетерохроматина) 1, 9, 15, 21
№ слайда 11

Описание слайда:
Общим для всех форм хромосомных болезней является множественность поражения. Это черепно-лицевые поражения, врожденные пороки развития систем органов, замедленные внутриутробные и постнатальные рост и развитие, отставание в психическом развитии, нарушения функций нервной, иммунной и эндокринной и других систем. Общим для всех форм хромосомных болезней является множественность поражения. Это черепно-лицевые поражения, врожденные пороки развития систем органов, замедленные внутриутробные и постнатальные рост и развитие, отставание в психическом развитии, нарушения функций нервной, иммунной и эндокринной и других систем. Степень поражения органов при хромосомных синдромах зависит от многих факторов — типа хромосомной аномалии, недостатка или избытка материала индивидуальной хромосомы, генотипа организма, условий среды, в котором развивается организм.
№ слайда 12

Описание слайда:
Женский кариотип, 46,ХХ
№ слайда 13

Описание слайда:
Мужской кариотип, 46,ХУ
№ слайда 14
. Для больных")
Описание слайда:
Примеры трисомий у человека Синдром Дауна (трисомия хромосомы №21). Для больных характерно: округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо, эпикант, запавшая спинка носа, косой (монголоидный) разрез глазных щелей, толстые губы, утолщенный язык с глубокими бороздами, выступающий изо рта. Из пороков внутренних органов наиболее типичны пороки сердца (дефекты межжелудочковой или межпредсердной перегородок) и органов пищеварения (атрезия двенадцатиперстной кишки).
№ слайда 15

Описание слайда:
Кариотип мальчика с трисомией хромосомы №21
№ слайда 16

Описание слайда:
Синдром Дауна Среди больных с синдромом Дауна с более высокой частотой, чем в популяции, встречаются случаи лейкемии и гипотиреоза. У маленьких детей резко выражена мышечная гипотония, а у детей старшего возраста часто обнаруживается катаракта. С самого раннего возраста отмечается отставание в умственном развитии. Среднее значение IQ составляет 50, но чаще встречается умеренная задержка умственного развития. Средняя продолжительность жизни при синдроме Дауна значительно ниже (36 лет), чем в популяции.
№ слайда 17
.")
Описание слайда:
Синдром Дауна СД — самая частая форма хромосомной патологии у человека (1:750). Цитогенетически синдром Дауна представлен простой трисомией (94% случаев), транслокационной формой (4%) или мозаицизмом (2% случаев). У мальчиков и девочек патология встречается одинаково часто. Достоверно установлено, что дети с синдромом Дауна чаще рождаются у пожилых родителей. Если возраст матери 35-46 лет, то вероятность рождения больного ребенка возрастает до 4,1%.
№ слайда 18

Описание слайда:
Зависимость частоты рождения детей с хромосомными болезнями от возраста матери
№ слайда 19

Описание слайда:
Робертсоновское слияние 2-х хромосом №21 в кариотипе больной с.Дауна
№ слайда 20
. Наблюдается с частотой 1 на 6000 родов.")
Описание слайда:
Синдром Патау (трисомия хромосомы №13). Наблюдается с частотой 1 на 6000 родов. У новорождённых средняя масса тела. Умственная отсталость тяжёлая, многие дети страдают глухотой. Нередко обнаруживается умеренная микроцефалия с нависающим лбом, большие анатомические дефекты мозга, незаращение нёба и верхней губы. Часто наблюдаются поперечная ладонная складка, полидактилия и узкие выпуклые ногти на пальцах рук. Более чем в 80% случаев регистрируются врождённые пороки сердца. Большинство больных (70%) настолько серьезно поражены, что погибают в возрасте до 6 месяцев, только 20% доживает до года.
№ слайда 21
. Дополнительная 18 хромосома обнаруж")
Описание слайда:
Синдром Эдвардса (трисомия по 18 хромосоме). Дополнительная 18 хромосома обнаруживается у 1 из 7000 новорождённых. Часто проявляется микроцефалия, низко посаженные уродливые уши и расщелина губы или нёба. Достаточно часто наблюдается отсутствие складки на мизинце, особый характер расположения кожных гребней на кончиках пальцев. Нередко наблюдается укорочение или даже отсутствие большого пальца на ногах, косолапость, синдактилия. Могут иметь место пороки сердца и крупных сосудов, врождённые аномалии лёгких, диафрагмы, почек и мочеточников, грыжи, складки на шее. Больные живут несколько месяцев, а у тех, кто всё же выживает, выявляется тяжелая умственная отсталость.
№ слайда 22

Описание слайда:
Трисомия 8 характерны отклонения в строении лица, пороки опорно-двигательного аппарата и мочевой системы. При клиническом обследовании выявляются выступающий лоб, косоглазие, эпикант, глубоко посаженные глаза, гипертелоризм глаз и сосков, высокое нёбо (иногда расщелина), толстые губы, вывернутая нижняя губа, большие ушные раковины. прогноз физического, психического развития и жизни неблагоприятный, хотя описаны пациенты в возрасте 17 лет.
№ слайда 23

Описание слайда:
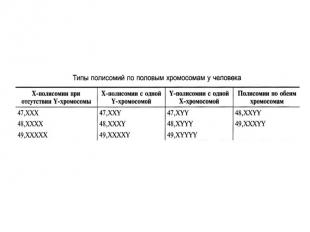
Аномалии сочетания половых хромосом Пол будущего ребенка определяется в момент оплодотворения в зависимости от сочетания половых хромосом (XX — женский организм, XY — мужской). При нарушении течения митоза могут образовываться необычные особи — гинандроморфы. Содержание половых хромосом в разных клетках таких особей может быть разное (мозаицизм). У человека могут быть разные случаи мозаицизма: ХХ/ХХХ, XY/XXY, ХО/ХХХ, XO/XXY и др.
№ слайда 24
№ слайда 25

Описание слайда:
Трипло-Х-женщины Частота встречаемости 1:1000. Кариотип 47,ХХХ. В настоящее время имеются описания тетра- и пентосомий X. Женский организм с мужеподобным телосложением. Могут быть недоразвиты первичные и вторичные половые признаки. В 75% случаев у больных наблюдается умеренная степень умственной отсталости. У некоторых из них нарушена функция яичников. Иногда такие женщины могут иметь детей. Повышен риск заболевания шизофренией. С увеличением числа дополнительных Х-хромосом нарастает степень отклонения от нормы.
№ слайда 26
Описание слайда:
Варианты синдрома Х-полисомии без Y-хромосомы с числом, большим, чем 3, встречаются редко. Варианты синдрома Х-полисомии без Y-хромосомы с числом, большим, чем 3, встречаются редко. С увеличением числа дополнительных Х-хромосом возрастает степень отклонений от нормы. У женщин с тетра- и пентасомией описаны отклонения в умственном развитии, черепно-лицевые дизморфии, аномалии зубов, скелета и половых органов. Однако женщины даже с тетрасомией по Х-хромосоме способны иметь потомство.
№ слайда 27

Описание слайда:
Синдром Шерешевского- Тернера моносомия X-хромосомы. Частота встречаемости 1:2000-1:3000. Кариотип 45,Х0. Отмечаются признаки дисплазии: короткая шея с избытком кожи и крыловидными складками, лимфатический отек стоп, голеней, кистей рук и предплечий, множественные пигментные пятна, низкорослость(рост взрослых 135-145см). Характерно недоразвитие первичных и вторичных половых признаков, дисгенезия гонад, сопровождающаяся первичной аменореей — бесплодие.
№ слайда 28

Описание слайда:
Синдром Клайнфельтера Кариотип 47,XXY, фенотип мужской. Частота — 1:1500 новорожденных мальчиков. Больных отличают следующие признаки: высокий рост, непропорционально длинные конечности, женский тип телосложения, гинекомастия, крипторхизм. Слабо развит волосяной покров, снижен интеллект. Бесплодие в результате недоразвития семенников. Инфантильность и поведенческие проблемы создают трудности в социальной адаптации.
№ слайда 29

№ слайда 30

Описание слайда:
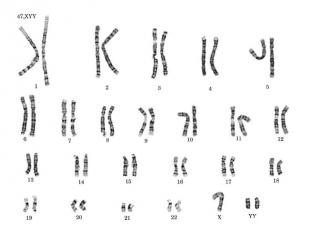
ХУУ — синдром характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г. Частота синдрома составляет среди новорожденных мальчиков около 1:1000. Наиболее частым признаком является высокий рост, который у взрослых больных составляет в среднем 186 см. У части больных отмечаются нерезко выраженные евнухоидные черты телосложения и диспластические признаки. Наличие добавочной Y-хромосомы может и не сопровождаться клинической патологией, но, несомненно, оно коррелирует как с интеллектуальным недоразвитием, так и с эмоционально-волевыми нарушениями.
№ слайда 31
№ слайда 32

Описание слайда:
Синдромы частичных анеуплоидий Помимо полных трисомий и моносомий известны синдромы, связанные с частичными трисомиями и моносомиями практически по любой хромосоме. Однако эти синдромы встречаются реже одного случая на 100 000 рождений.
№ слайда 33

Описание слайда:
Синдром трисомии по короткому плечу 9-й хромосомы Для больных с трисомией 9р+ характерны умственная отсталость, задержка роста, микроцефалия, антимонголоидный разрез глазных щелей, глубоко посаженные глаза, опущенные уголки рта, нос с характерным округлым кончиком, низко расположенные оттопыренные ушные раковины, недоразвитие ногтей и дистальных фаланг пальцев рук. Часто наблюдаются выступающие лобные кости, повышенная обволошенность, пятна цвета кофе с молоком на коже, эпикант, косоглазие, высокое дуто-образное нёбо, короткая шея, сколиоз, частичная синдактилия пальцев стоп. Примерно в четверти случаев обнаруживаются врожденные пороки сердца. Прогноз для жизни сравнительно благоприятный (при отсутствии патологии внутренних органов) — описаны больные, достигшие преклонного возраста. По частоте встречаемости среди детей-олигофренов занимает 2 место после болезни Дауна.
№ слайда 34
. Основные клинико-мор")
Описание слайда:
Синдром кольцевой хромосомы 9 Кариотип 46 ХХ или ХУ, r (9). Основные клинико-морфологические признаки: характерное лицо -микроцефалия, косой разрез глаз, ретрогнатия, короткая шея. У всех больных наблюдается умственная отсталость, задержка психомоторного развития.
№ слайда 35

Описание слайда:
Делеция дистальной части короткого плеча хромосомы 5 у человека Для данного синдрома наиболее характерны специфический плач, напоминающий кошачье мяуканье, лунообразное лицо, мышечная гипотония, умственное и физическое недоразвитие, микроцефалия, низко расположенные, иногда деформированные ушные раковины, эпикант, антимонголоидный разрез глазных щелей, косоглазие.
№ слайда 36
 Популяционная частота - 1:100")
Описание слайда:
Синдром Вольфа-Хиршхорна (частичная моносомия 4р-) Популяционная частота — 1:100000. обусловлен делецией сегмента короткого плеча хромосомы 4. Клинически характеризуется мВПР (микроцефалия, клювовидный нос, гипертелоризм, эпикант, аномальные ушные раковины, расщелины верхней губы и нёба, аномалии глазных яблок, антимонголоидный разрез глаз, маленький рот, пороки внутренних органов) с последующей резкой задержкой физического и психомоторного развития. Жизнеспособность детей резко снижена. Большинство умирают в возрасте до 1 года.
№ слайда 37
 обусловлен делецией длинного плеча 13-й хромосомы. Популяц")
Описание слайда:
Синдром Орбели (13q-) обусловлен делецией длинного плеча 13-й хромосомы. Популяционная частота не установлена. Характерны микроцефалия, отсутствие носовой вырезки (лоб непосредственно переходит в нос), эпикант, антимонголоидный разрез глаз, высокое нёбо, низко расположенные деформированные ушные раковины. Отмечаются поражения глаз (микрофтальмия, косоглазие, катаракта, ретинобластома), опорно-двигательного аппарата (короткая шея, синдактилии кистей и стоп), атрезии прямой кишки. Часты пороки развития сердца, почек, головного мозга. Для всех детей с синдромом Орбели характерна глубокая олигофрения. Большинство больных с синдромом 13q- погибают на 1-м году жизни.
№ слайда 38

Описание слайда:
Синдром Прадера-Вилли у 70% больных наблюдается частичная делеция длинного плеча 15-й хромосомы (отцовская аллель), у 5% заболевание связано с другими перестройками хромосомы 15. Характерные внешние признаки: череп со сдавленной с боков лобной частью, миндалевидный разрез глаз, опущенные углы рта, маленькие стопы и кисти) Наблюдается отставание умственного развития, поведенческие нарушения, задержка физического развития, низкорослость, гипотония, гипогонадизм.
№ слайда 39

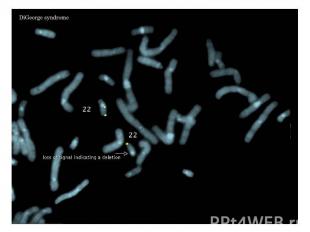
Описание слайда:
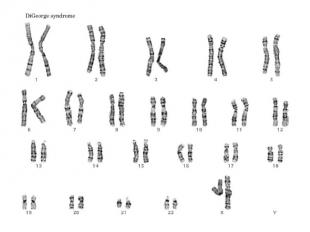
Синдром Ди Джорджи Частичная моносомия 22q11.2. Популяционная частота — 1:20 000. Больные имеют следующие клинические признаки: дефекты развития третьего и четвертого глоточных карманов, что приводит к гипоплазии или аплазии тимуса с дефицитом Т-клеток и иммунодефицитом; дефекты сердца и характерные дисморфичные изменения лица (гипертелоризм, короткая ось век, эпикант, широкий короткий нос с вывернутой носовой пластиной, слабо обозначенный рот, микроретрогнатия, низко расположенные дисморфичные уши),волчья пасть. Дети с синдромом Ди Джорджи часто физически и умственно отсталые.
№ слайда 40
№ слайда 41
№ слайда 42

Описание слайда:
синдром Мартина — Белла синдром ломкой, или фрагильной Х-хромосомы. Название синдрома объясняется особой формой строения Х-хромосомы, которая имеет хорошо заметную перетяжку на конце длинного плеча. является одной из наиболее распространенных причин наследственных олигофрений у мужчин (примерно 1 на 2000 рождений).
№ слайда 43

Описание слайда:
синдром Мартина — Белла для больных характерны некоторые морфологические признаки, которые не всегда отчетливо проявляются (высокий выпуклый лоб, крупные уши и челюсти, крупные кисти рук, увеличенные яички). Умственное развитие колеблется между значениями IQ от 30 до 65 (иногда в границах нормы). Речь изобилует повторами, часто встречается своеобразное заикание. Для детей характерна двигательная расторможенность и некоторые симптомы аутизма (ребенок избегает глазного контакта, производит стереотипные движения руками, испытывает страхи). Даже при легкой степени интеллектуальной недостаточности дети с трудом овладевают навыками счета и письма.
№ слайда 44

Описание слайда:
синдром Мартина — Белла Генетический механизм заболевания связан с экспансией тринуклеотидных повторов (CGG — цитозин-гуанин-гуанин) в соответствующем перетяжке участке Х-хромосомы. В норме число повторов не должно превышать 50. Количество повторов от 50 до 200 считается премутацией, выраженная картина болезни наблюдается при наличии более 200 повторов. Для данного заболевания характерно явление антиципации, т.е. усиление тяжести заболевания от поколения к поколению. Это связано с нарастанием числа тринуклеотидных повторов в мутировавшем участке Х-хромосомы.
Источник
- Скачать презентацию (2.61 Мб)
- 46 загрузок
- 4.9 оценка
Ваша оценка презентации
Оцените презентацию по шкале от 1 до 5 баллов
Рецензии
Добавить свою рецензию
Аннотация к презентации
Презентация на тему «Генетические синдромы» подготовлена для показа на уроках биологии, посвященных изучению болезней. Данный информационный презентационный материал поможет выступающему познакомить школьников с генетическими синдромами. Разработка состоит из 32 слайдов.
Краткое содержание
- Синдром Дальтона
- Гемофилия
- Синдром Дауна
- Ихтиоз
- Прогерия
- Циклопия
- Альбинизм
- Синдром Патау
- Синдром Марфана
Содержание
Слайд 1
ГЕНЕТИЧЕСКИЕ СИНДРОМЫ
ПРЕЗЕНТАЦИЮ ПОДГОТОВИЛА УЧИТЕЛЬ БИОЛОГИИ
МОУ «СОШ №24» КИРОВСКОГО РАЙОНА Г.САРАТОВА
ЧАЛОВА ГАЛИНА ЕВГЕНЬЕВНА 2012-2013 УЧЕБНЫЙ ГОД
Слайд 2
СИНДРОМ ДАЛЬТОНА
Люди с нормальным цветным зрением имеют в колбочках три пигмента (красный, зелёный и синий),их называют трихроматами. Джон Дальтон был протанопом (не различал красный цвет), он не знал о своей цветовой слепоте до 26 лет, синдром описал в 1794 году. Передача дальтонизма по наследству связана с X-хромосомой и практически всегда передаётся от матери-носителя гена к сыну, в результате чего в двадцать раз чаще проявляется у мужчин имеющих набор половых хромосом XY. Разной степенью дальтонизма страдают 2—8 % мужчин и 0,4 % женщин. Согласно исследованиям британских учёных, люди, которым трудно различать красные и зелёные цвета, могут различать множество других оттенков, в частности, оттенки цвета хаки.
Слайд 3
ГЕМОФИЛИЯ
Гемофилия — наследственное заболевание(1:10000), обусловленное недостаточностью системы свертывания крови и проявляющееся кровоточивостью. Болеют главным образом мужчины, женщины — лишь носители мутантного гена и передают гемофилию сыновьям. Королева Виктория передала ген гемофилии царевичу Алексею.
Слайд 4
СИНДРОМ ДАУНА
Во все времена рождались дети с синдромом Дауна, но впервые этот синдром описал английский врач Джон Даун в 1886 году. А уже тайну этого синдрома – лишней сорок седьмой хромосомы открыл в 1959 году французский генетик ЖеромЛеженом. Лишняя хромосома является результатом нарушения созревания половых клеток. Эта патология не является редкостью
Слайд 5
ИХТИОЗ
Ихтиоз (крокодилова кожа, змеиная кожа, рыбья чешуя) – достаточно редкая, проявляющаяся еще в раннем детстве и протекающая хронически наследственная аномалия ороговения кожи(1:20000)
Слайд 6
СИНДРОМ ВЕРНЕРА (ПРОГЕРИЯ)
Прогерия– аутосомно-рецессивное заболевание характеризующееся проявлением симптомов преждевременного старения кожи, сосудистой и репродуктивной системы, костей из-за нарушений в гене 8 хромосомы.
Слайд 7
ЦИКЛОПИЯ
В индийском городеЧеннайродился ребенок с одним глазом. Девочка появиласьс редким генетическим отклонением -циклопией. У ребенка не было носа и только один глаз, расположенный в центре лба. Кроме того, ее мозг слился в одно полушарие. Циклопия случается частотой — один раз на миллион новорожденных. Это отклонение может быть вызвано недостатком холестерина или диабетом у матери , внешней причиной, мутациеи.
Слайд 8
АЛЬБИНИЗМ
Альбинизм(1-20000) — это отсутствие пигмента в коже, волосах, тканях глаза.В основе альбинизма лежит нарушение образования в клетках кожи, волосяных луковицах и в глазу черного пигмента — меланина. Обычно этот пигмент образуется из вещества, которое называется тирозин, под воздействием фермента тирозиназы. Это сложный биохимический процесс, в котором участвует много различных веществ и ферментов. При альбинизме имеется дефект в генах, регулирующих этот процесс. Опубликовано около 700 родословных семей, члены которых страдали альбинизмом. Наследственные заболевания имеют тенденцию распространяться в небольших этнических группах в связи с большой частотой родственных браков. Выявлены очаги альбинизма в Северной Ирландии. В Южной Панаме среди племени карибе куна были обнаружены сотни альбиносов. Тропическое солнце очень сильно обжигало альбинотическую кожу и слепило глаза, поэтому альбиносы в этом районе вели ночной образ жизни. Их прозвали «детьми Луны».
Слайд 9
СИНДРОМ ПАТАУ
Синдром Патау( трисомия по хромосоме 13) –встречается 1:5000-7000, описан в 1960году.Эта аномалия вызывает расщепление губы и неба («заячья губа» и «волчья пасть»),пороки развития головного мозга, глазных яблок , полидактилию. Выживает 10% детей
Слайд 10
СИНДРОМ МАРФАНА
Синдром Марфана- генетическое заболевание, связанное с поражением соединительной ткани, проявляющееся очень худым телосложением, длинными руками и ногами, худыми тонкими, длинными пальцами, поражениями сердечно-сосудистой системы (пороки клапанов и аорты). Могут поражаться органы: глаза, легкие, кости, мозговые оболочки. Синдром Марфана развивается вследствие дефекта в гене, отвечающего за структуру белка соединительной ткани (фибрилина)
Слайд 11
СИНДРОМ ЛЕЖЕНА
Болезнь Лежена(1:3000) -это наследственное генетическое заболевание, связанное с изменением строения 5-ой хромосомы. Болезнь получила название по имени французского ученогоЖеромаЛежена в 1963 году. Дети рождаются с массой 2,5 кг, лунообразным лицом, косым разрезом глаз, эпикантусом, микроцефалией, короткой шеей, умственно неполноценные. Дети с особым строением гортани, небольшим надгортанником , поэтому для этого заболевания характерен необычный детский крик, напоминающий кошачий.
Слайд 12
СИНДРОМ ЖИЛЬБЕРА
Болезнь Жильбера– это наследственное заболевание, которое проявляется повышением уровня билирубина в крови, желтухой Причиной болезни является генетически обусловленный недостаток специального фермента печени (глюкуронилтрансферазы), который участвует в обмене билирубина. Повышенный уровень билирубина при синдромеЖильбераотмечается с самого рождения. Симптомы болезни Жильбера непостоянны и появляются в результате стресса, физической нагрузки , голодания, приема алкоголя, лекарств, после вирусных заболеваний (грипп)
Слайд 13
СИНДРОМ ВИЛЬСОНА
Болезнь Вильсона – наследственное расстройство обмена меди, приводящее к избыточному отложению последней во внутренних органах (печени, роговице, головном мозге ). Основой заболевания является нарушение выделения меди с желчью. Вследствие избыточного накопления меди повышается продукция свободных радикалов и повреждаются ткани. Ген болезни Вильсона, расположенный в 13-й хромосоме, кодирует структуру белка, транспортирующего медь.
Слайд 14
СИНДРОМ АЛЬБЕРС-ШЕНБЕРГА
Мраморная болезнь -врожденный остеосклероз, редкое заболевание почти всех костей скелета. Открыт синдром в 1904 году немецким хирургом Альберс- Шенбергом. Описано в настоящее время 300 случаев болезни. Симптомы мраморной болезни:
— Быстрая утомляемость при ходьбе- Боли в конечностях- Развитие патологических переломов костей- Развитие деформаций костей конечностей- Увеличение селезенки, печени и лимфатических узлов- Повышение внутриглазного давления и понижение слухаУ новорожденных с мраморной болезнью уже имеется ,или полная слепота, или частичная слепота ,вследствие атрофии зрительных нервов.
Слайд 15
СИНДРОМ РЕКЛИНГХАУЗЕНА
Нейрофиброматоз– наследственное ,семейное заболевание при котором на коже появляется множество мясистых мягких на ощупь опухолей, которые состоят из видоизмененной нервной ткани (нейрофибром).Это заболевание может проявиться в первые десять лет жизни. Мужчины болеют им примерно в два раза чаще ,чем женщины Тип наследования аутосомно-доминантный. Частота возникновения заболевания наблюдается примерно у каждого 3500 новорождённого. Риск наследования ребёнком данной патологии при наличии у одного из родителей равен 50%, у обоих — 66,7%. Ген картирован в 17-й хромосоме.
Слайд 16
СИНДРОМ НОТТА
Болезнь Нотта (пружинящий палец, щелкающий палец, стенозирующийлигаментит сгибателей пальцев кисти) – достаточно распространенное заболевание сухожилий сгибателей пальцев и окружающих их связок. На начальном этапе заболевания разгибание пальца еще возможно, однако оно сопровождается характерным щелчком (отсюда и пошло название “щелкающий палец»). По мере течения болезниНоттаразг?