Синдромы клайнфельтера тернера у животных

Синдром Клайнфельтера и Шерешевского — Тернера. Гермафродитизм
Синдром Клайнфельтера наблюдается у лиц с мужским фенотипом. Характерной особенностью, объединяющей индивидуумов с различным набором половых хромосом в одну группу, является недоразвитие яичек и отсутствие сперматогенеза. Больные с синдромом Клайнфельтера обычно имеют высокий рост, евнухоидные пропорции тела (узкие плечи, широкий таз), склонность к ожирению, иногда гинекомастию (одно- или двустороннюю), оволосение по женскому типу. Основная масса таких больных имеет кариотип XXY.
Описаны также больные с набором половых хромосом XXXY, XXXXY, XXYY и др. При исследовании интерфазных ядер у таких больных обнаруживают как Х-, так и Y-хроматин, причем число телец Х-хроматина равно числу Х-хромосом минус единица. Так, например, у индивидуума с кариотипом XXXXY в ядрах некоторых клеток может быть обнаружено до 3 телец Х-хроматина и одно Y-хроматина.
Синдром Шерешевского — Тернера наблюдается у лиц фенотнпически женского пола с задержкой роста и полового развития, нормальными наружными и недоразвитыми внутренними половыми органами. Наиболее характерными чертами являются отсутствие гонад, первичная аменорея, бесплодие, короткая шея и крыловидные складки кожи на шее.
Женщины с синдромом Тернера имеют кариотип ХО, Х-хроматин в клетках их тканей отсутствует.
Ложный мужской гермафродитизм. У больных имеются тестикулы, а дифференциация других внутренних и всех или некоторых наружных половых органов отклоняется в женскую сторону. Разновидностью этого состояния является аномалия полового развития, называемая тестикулярной феминизацией, проявляющаяся присутствием тестикул у фенотипических женщин с первичной аменореей. Отмечается нормальное, часто чрезмерное развитие грудн. Наружные половые органы женские, внутренние отсутствуют. Кариотип этих больных содержит XY-хромосомы.
В ядрах клеток Х-хроматин отсутствует, а Y-хроматин обнаруживается.
Ложный женский гермафродитизм — аномалия полового развития, несвязанная с нарушением в системе половых хромосом. Заболевание обусловлено врожденной гиперплазией коры надпочечников. Основные клинические проявления синдрома вызываются избытком мужских гормонов — андрогенов, обусловливающих вирилизирующий эффект. Все больные имеют Х-хроматин и нормальный женский кариотип.
Истинный гермафродитизм. Под этим названием понимают такое интерсексуальное состояние, когда у индивидуума одновременно имеются две половые железы — мужского и женского пола (яичко и яичник) или железы, состоящие из тканей мужской и женской половых желез (овотестис). Строение наружных половых органов характеризуется различной степенью переходов от одного пола к другому. Около 60% описанных случаев имели ХХ-хромосомную конституцию. В 40% случаев в кариотипе имелась и Y-хромосома.
Полисомия по Х-хромосоме у женщин (XXX, ХХХХ, ХХХХХ) характеризуется наличием в некоторых интерфазных ядрах двух — трех телец Х-хроматнна. Это пограничное между патологией и нормой состояние сопровождается эндокринным дисбалансом и в первую очередь нарушением функции яичников. У части обследованных женщин выявлены бесплодие, нерегулярность менструального цикла, преждевременный климакс.
Аномальные наборы половых хромосом могут возникать не только в процессе гамстогенеза (при нарушении распределения их в митозе), но и в процессе эмбрионального развития (при нарушении распределения их в анафазе митотического деления). Последние нарушения приводят к появлению линий (клонов) клеток, отличающихся от исходных; формируется мозаичный (по половым хромосомам) организм.
Например, 46, XY/47, XXY — наиболее частый тип мозаичности по половым хромосомам у мужчин, и 45, Х/46, XX — у женщин. Процентное содержание клеток разных клонов, распределение их в тканях организма зависит от стадии эмбрионального развития, на которой появилась аномалия, и от выживаемости аномального клона. В зависимости от преобладания клеток того или иного клона варьируют и клинические проявления.
Истинную природу интерсексуальных заболеваний можно выяснить только при сопоставлении результатов клинического осмотра и лабораторных исследований. Большую ценность для диагностики представляет установление кариотипа больного и комплекса половых хромосом. Число и состав половых хромосом можно определить путем исследования Х- и Y-хроматина, так как между числом половых хромосом и числом телец полового хроматина в покоящемся ядре существует вполне определенное соотношение.
— Также рекомендуем «Определение половой принадлежности материалов. Половая принадлежность сред организма»
Оглавление темы «Определение половой принадлежности клеток»:
1. Содержание Y-хроматина в клетках. Аномалии содержания полового хроматина
2. Синдром Клайнфельтера и Шерешевского — Тернера. Гермафродитизм
3. Определение половой принадлежности материалов. Половая принадлежность сред организма
4. Половая принадлежность по Х-хроматину. Определение пола по Х-хроматину
5. Приготовление препаратов крови для выявления Х-хроматина. Выявление пола по крови и слюны
6. Микроскопия для определения пола. Исследование препаратов для определения пола клеток
7. Микроскопия крови, волос для определения пола. Оценка результатов выявления пола
8. Статистический метод определения пола. Последовательный анализ Вальда
9. Половая принадлежность Y-хроматину. Приготовление препаратов для определения пола по Y-хроматину
10. Микроскопия для выявления Y-хроматина. Исследование препаратов на Y-хроматин
Источник
В настоящее время важное место в сфере медицинских наук занимает медицинская генетика, которая изучает генетические заболевания. Вы уже знаете, что
вариантов мутаций множество: от выпадения отдельных нуклеотидов в гене до утраты целых хромосом. Количество вариантов мутаций и их сочетаний — бесчисленно, что делает медицинскую генетику неисчерпаемой.
Медицинская генетика играет важную роль при планировании семьи, служит для предупреждения наследственных заболеваний. В данной статье мы изучим некоторые наиболее известные наследственные заболевания.

Альбинизм
Альбинизм (лат. albus — белый) — врожденное заболевание, наследуемое по рецессивному типу и связанное с нарушением синтеза
черного пигмента — меланина (греч. melanos – черный) у животных или хлорофилла (у растений). Альбинизм возникает в результате
генной мутации в участке ДНК, ответственном за синтез меланина/хлорофилла.
Растения с утратой хлорофилла утрачивают способность улавливать солнечный свет, поэтому полный альбинизм для них
заканчивается летально. У животных мутация происходит в гене тирозиназы, в связи с чем меланин не синтезируется: кожа
альбиносов не способна загорать, для них характерен больший риск ожогов и рака кожи.
Радужка пропускает свет и становится красноватого оттенка, за счет кровеносных сосудов, расположенных на глазном дне.

Серповидно-клеточная анемия
Это наследственное заболевание, вызванное генной мутацией, в результате которой меняется конформация молекулы гемоглобина:
эритроцит становится выгнутым и напоминает серп.
Эта болезнь встречается особенно часто в странах, эндемичных по малярии. Больные серповидно-клеточной анемией обладают
повышенной устойчивостью к заражению малярийным плазмодием, поэтому эту болезнь можно рассматривать как результат действия
естественного отбора: с ней выживаемость людей повышалась, и они продолжали род, передавая мутацию потомкам.

Синдром Дауна
Наследственное заболевание, возникающее в результате геномной патологии: трисомия по 21-ой паре хромосом. Это означает, что
вместо двух хромосом в 21-ой паре появляется одна лишняя — третья хромосома. Причина ее появления связана с нерасхождением
хромосом во время мейоза.
Риск рождения ребенка с синдромом Дауна возрастает с увеличением возраста матери.

Синдром проявляется характерными признаками: плоское лицо, приоткрытый рот, поперечная ладонная складка, гиперподвижность суставов,
эпикантус (кожная складка, прикрывающая угол глазной щели).

Синдром Эдвардса
Наследственное заболевание, вызванное геномной мутацией — трисомией по 18 паре хромосом. Причина — нерасхождение хромосом во время
мейоза, еще до оплодотворения. Чаще болезнь встречается у пожилых матерей.
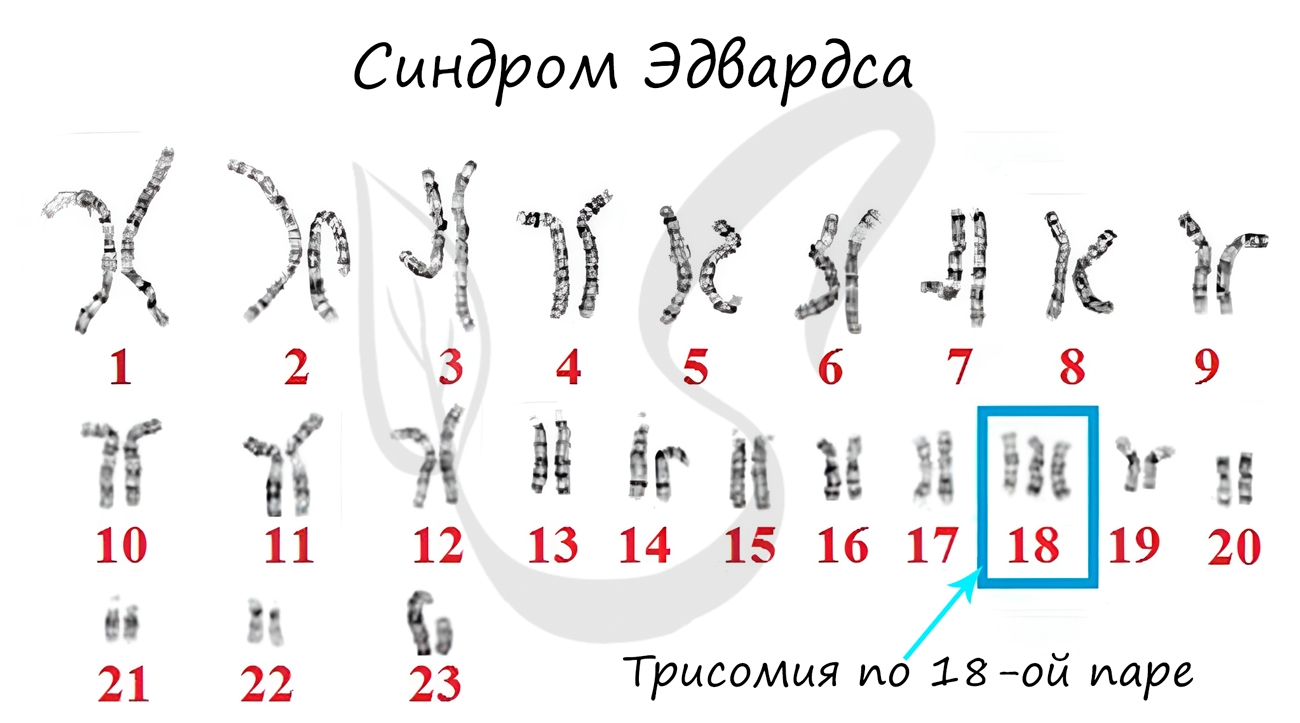
Детям с синдромом Эдвардса сопутствуют пороки сердца и сосудов: 60% детей умирают в течение первых 3 месяцев, до 1 года доживают лишь 5-10% детей.
Синдром Патау
Наследственное заболевание, обусловленной геномной мутацией — трисомией по 13 паре хромосом. Существует зависимость между возрастом матери
и вероятностью рождения ребенка с синдромом Патау (с возрастом риск увеличивается), хотя зависимость менее выражена, чем в случае с синдромом
Дауна.
При данном синдроме обнаруживаются тяжелые врожденные пороки сердца и сосудов, нервной системы. Большинство детей с синдромом Патау умирают
в первые недели или месяцы жизни, до 1 года доживают лишь 5% детей.

Синдром Клайнфельтера
Синдром Клайнфельтера представляет собой наследственное заболевание, развивающееся вследствие полисомии по X и Y хромосомам (половым
хромосомам). Возможны несколько вариантов генотипов: XXY (самый частый), XYY, XXXY, XXXXY, XXXYY. На всякий случай напомню норму
мужского генотипа — «XY» 🙂
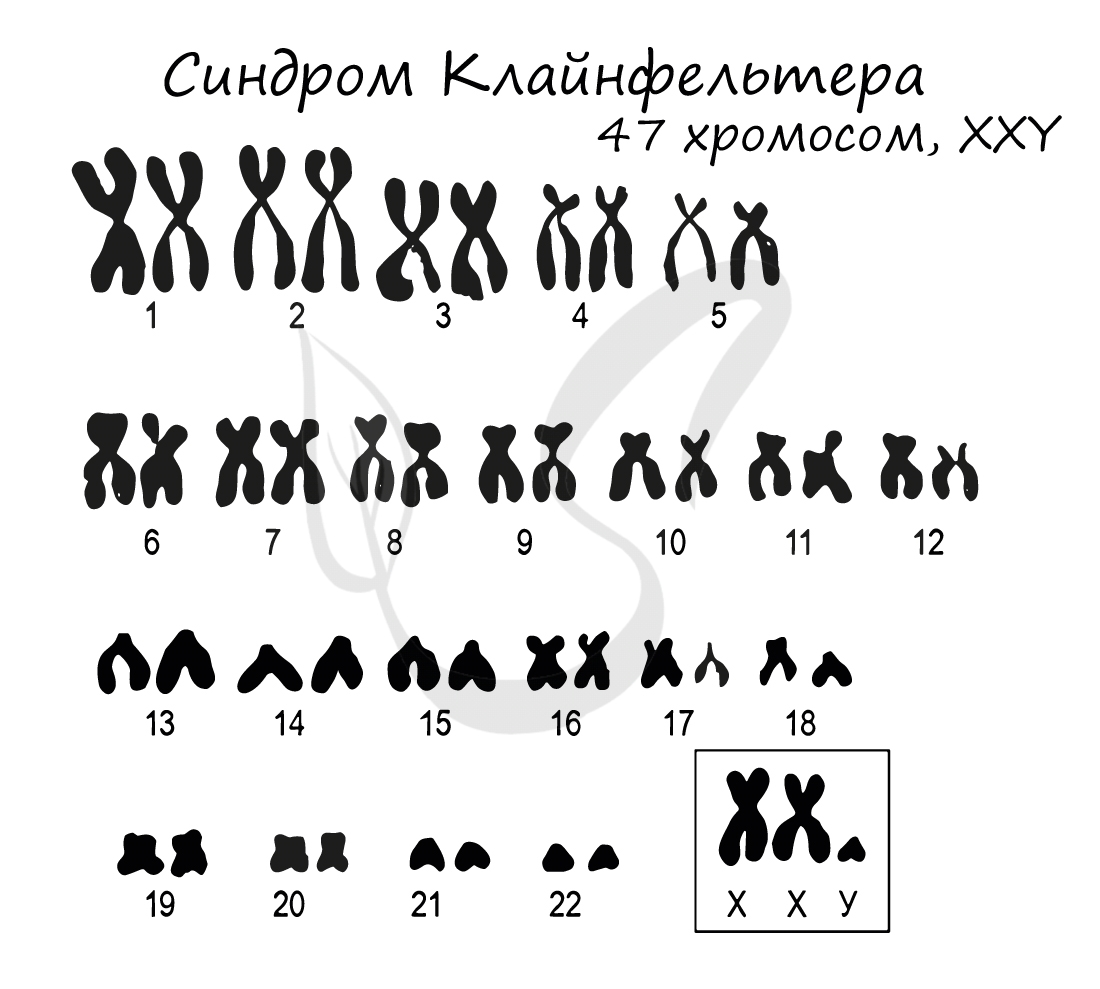
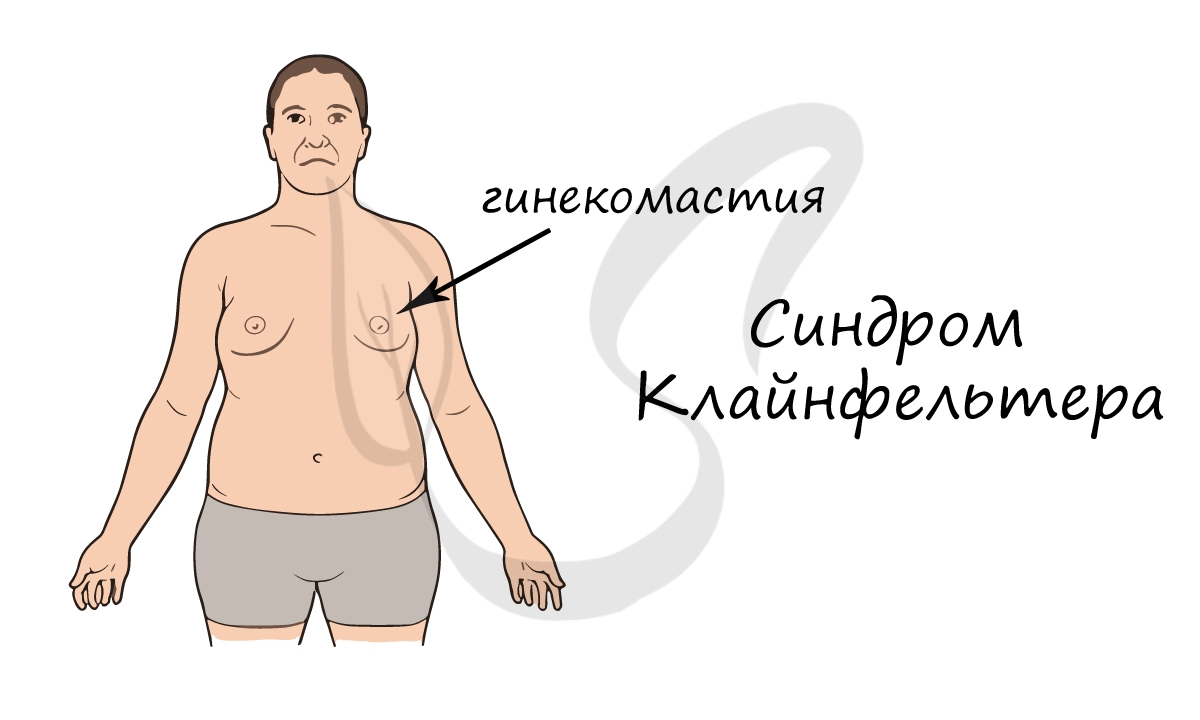
Диагностируется синдром относительно поздно, так как проявляется только после полового созревания. В подростковом возрасте развивается гинекомастия
(увеличение грудной железы), сохраняющаяся всю жизнь. Характерно наличие маленьких плотных яичек. Синдром Клайнфельтера приводит к бесплодию.
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера — наследственное заболевание, характерное только для женщин и возникающее в результате моносомии по половым
хромосомам. Генотип человека при таком заболевании будет записан как X0 (45 хромосом).
Больные синдромом Шерешевского-Тернера низкорослые, инфантильные, их психический статус характеризуется состоянием беспричинно приподнятого
настроения — эйфорией. Тем не менее, интеллект и жизнеспособность сохранены. Из-за геномной мутации (X0) стерильны.

© Беллевич Юрий Сергеевич 2018-2020
Данная статья написана Беллевичем Юрием Сергеевичем и является его интеллектуальной собственностью. Копирование, распространение
(в том числе путем копирования на другие сайты и ресурсы в Интернете) или любое иное использование информации и объектов
без предварительного согласия правообладателя преследуется по закону. Для получения материалов статьи и разрешения их использования,
обратитесь, пожалуйста, к Беллевичу Юрию.
Источник
БОЛЕЗНЬ КЛАЙНФЕЛЬТОРА. В 1942 г. Г. Клайнфельтер совместно с другими авторами описал своеобразный синдром мужского гипогонадизма.
Клинически он проявляется евнухоидными признаками: высоким ростом, недоразвитием вторичных половых признаков, высоким голосом, недостаточным развитием оволосения, гинекомастией, атрофией яичек, бесплодием.
Психически характеризуется умственно отсталостью и психической вялостью. Частота синдрома среди населения 1:108. Во вспомогательных школах 0,1% составляют учащиеся с синдромом Клайнфельтера от числа всех учащихся. Помимо умственной отсталости заболевание нередко сопровождается асоциальным поведением в юношеском возрасте.
Гистологически отмечается склерозирующая гиалинизация семенных канальцев, гиперплазия интерстициальных клеток Лейдига, гибель клеток Сертоли и повышенное выделение фолликулостимулирующего гормона.
В 1956 г. Р. Бриге и М. Барр провели исследование кариотипа и полового хроматина у больных и выявили лишнюю Х-хромосому в кариотипе. Таким образом, кариотип этих больных представлен 47 хромосомами (44а + XX Y). В последующем было показано, что бывают случаи болезни Клайнфельтера с тремя и даже четырьмя X—хромосомами: 44а + XXX Y, 44а + XXXX Y. Чем больше выявлено Х-хромосом в кариотипе, тем более тяжело проявляется заболевание фенотипически. Но не у всех больных с болезнью Клайнфельтера определялась лишняя X-хромосома и половой хроматин. Наблюдались больные, у которых был нормальный кариотип. Поэтому в настоящее время выделяют болезнь Клайнфельтера, когда обнаруживается лишняя Х-хромосома или несколько, и синдром Клайнфельтера в тех случаях, где кариотип нормальный.
При синдроме Клайнфельтера клиника заболевания проявляется менее выраженно, чем при болезни Клайнфельтера. При болезни Клайнфельтера сперматогенез не обнаружен, а при синдроме Клайнфельтера обнаруживаются семенные канальцы со сперматогенезом. Больные болезнью Клайнфельтера из-за аспермии бесплодны. В последующем было показано, что примерно 11 % случаев мужского-бесплодия обусловлено болезнью Клайнфельтера.
Больные с болезнью-Клайнфельтера страдают дебильностью разной степени выраженности. Большой процент этих больных встречается среди олигофренов.
Болезнь Клайнфельтера может сочетаться с атрофической миотонией, мозжечковой атаксией, болезнью Дауна.
Этиология этого заболевания не выяснена. Полагают, что имеет значение возраст матери при рождении ребенка. Чем она старше, тем больше опасность рождения больного ребенка. У матери кариотип нормальный, но при гаметогенезе в одну яйцеклетку может попасть две Х-хромосомы. При оплодотворении такой яйцеклетки нормальным спермием рождается ребенок с болезнью Клайнфельтера. Но хромосомная аберрация может произойти и в результате мутации в начале деления зиготы. Это может быть и у молодых матерей. Наследственность при болезни Клайнфельтера не изучена, так как эти больные не оставляют потомства.
Патогенез этого заболевания объясняется активностью лишних Х-хромосом на ранних стадиях развития зародыша, когда происходит развитие гонад, вследствие гибели клеток Сертоли и угнетения выработки ингибина (Х-гормона) тормозящее действие его на гипофиз снижается, вследствие чего выработка им гонадотропинов резко возрастает. Слабоумие обусловлено нарушением генного баланса из-за лишней хромосомы.
Эффективного лечения нет. Применение андрогенов из-за бесплодия к желаемому эффекту не приводит, но может привести к гиперсексуальности и половым эксцессам, так как эти больные олигофрены.
СИНДРОМ ШЕРЕШЕВСКОГО — ТЕРНЕРА. В 1925 г. Н. А. Шерешевский описал заболевание у 20-летней женщины, которое характеризовалось инфантилизмом, недоразвитием гонад, широкой складкой кожи на шее и другими симптомами. Причиной этого заболевания он считал недоразвитие передней доли гипофиза и яичников.
В 1938 г. Г. Тернер опубликовал работу о наблюдении им 7 женщин с подобным синдромом. Он также считал причиной его нарушение функции гипофиза. С этого времени это заболевание получило название синдрома Шерешевского — Тернера. В последующие годы у этих больных было выявлено повышенное выделение гонадотропного гормона, что исключало недостаточность функции гипофиза.
Новое направление в изучении синдрома Шерешевского — Тернера началось с 1949 г. после открытия М. Барром полового хроматина в клетках женских организмов. У женщин, больных синдромом Шерешевского— Тернера, не оказалось полового хроматина. Было высказано предположение, что у них генотип представлен ХY—хромосомами, но под влиянием каких-то причин фенотип пошел по женскому типу. У этих женщин встречаются заболевания, характерные для мужчин, сцепленные с половой хромосомой, такие как цветовая слепота, гемофилия, каорктация аорты и др.
В 1956 г. П. Полани предположил, что у этих женщин генотип не ХY, а Х0, то есть у них нет одной Х-хромосомы и кариотип представлен 45 хромосомами. Это надо было доказать. В 1959 г. это было подтверждено К. Е. Фордом. В дальнейшем по мере совершенствования цитогенетических методов исследования показали, что синдром Шерешевского — Тернера может быть вызван не только моносомией по Х-хромосоме, но и морфологическими ее изменениями (делеция короткого или длинного плеча, кольцевые хромосомы, фрагментация, изохромосомы и др.), наблюдали мозаицизм ХХ/ХО, гинандроморфический мозаицизм, когда одна половина тела женщины содержала нормальный набор хромосом — XX, а вторая — ХО.
Клинически синдром Шерешевского — Тернера представлен многообразными симптомами. Это женщины низкого роста (130—145 см) и реже немного выше. У них половой инфантилизм, недоразвиты наружные половые органы, узкое влагалище, недоразвита матка, гипертрофирован клитор, недоразвиты яичники. На месте их определяется соединительнотканный тяж, редко бывают фолликулы. Месячные отсутствуют или бывают однократные и скудные. Очень скудное оволосение на лобке или оно вовсе отсутствует. Грудные железы отсутствуют, иногда на их месте отмечается скопление жира. Соски недоразвиты, ареолы втянуты, широко расставлены и не пигментированы. Лицо таких больных кажется старше их возраста. Ушные раковины деформированы, расположены низко, шея короткая, волосы на шее растут очень низко, отмечается широкая кожная складка, идущая от сосцевидных отростков к надплечью. Нередко отмечается эпикантус, микро-и ретрогнатия. Твердое небо высокое и узкое.
На нижних конечностях отмечается ряд изменений. Ноги короткие, толстые. 3-й, 4-й и 5-й пальцы укорочены, искривлены и неправильно располагаются над стопой. На рентгенограмме трубчатых костей отмечается задержка окостенения, нет слияния эпифизов и метафизов даже у 25-летних больных, а рост их прекращается в 15—18 лет.
Со стороны внутренних органов отмечается стеноз аорты (каорктация), стеноз легочной артерии, незаращение межжелудочковой перегородки, аномалия почек (подковообразная почка), гипертония.
Со стороны нервной системы существенных изменений не определяется, интеллект у этих больных не страдает или нарушается мало с разной степенью умственной отсталости. У этих женщин отмечается повышенное выделение гонадотропина и резко снижено выделение эстрогенов.
Однако не все из указанных симптомов бывают одновременно у одного больного . Их может быть несколько.
При рождении ребенка не всегда можно установить диагноз. Но нередко у таких детей после рождения отмечается лимфатический отек конечностей и избыток кожи на шее, которая потом превращается в кожную складку. Дети часто рождаются недоношенными, с малым ростом. Но обычно диагноз заболевания устанавливается не сразу, а спустя годы, когда обнаруживается задержка роста и половой инфантилизм. Большое значение у этих детей имеет определение полового хроматина (он у них отсутствует), а также определение кариотипа — 45 хромосом при Х0 или мозаицизм и другие изменения Х-хромосомы.
Причины рождения детей с синдромом Шерешевского—Тернера до настоящего времени окончательно не ясны. Возраст родителей как будто не играет роли. Чаще такие дети рождаются у родителей низкого роста, хотя половой хроматин у них есть и не наблюдается хромосомных аберраций при обычном исследовании кариотипа. Потеря Х-хромосомы происходит, по-видимому, на первых этапах дробления зиготы. Без Х-хромосомы не происходит развития яичников, нарушается синтез структурных белков и ферментов, а это вызывает появление различных аномалий.
Путем определения группы крови XD — ген локализованный в коротком плече Х-хромосомы, у этих больных может быть как материнской, так и отцовской (в случаях, где родители различались по этому гену).
Эффективного лечения этих больных не существует. Назначаемое гормональное лечение направлено на увеличение роста и феминизации. К сожалению, оно назначается уже в возрасте после 12—14 лет, когда обнаруживается аменоррея и отставание в росте. Такое лечение приводило к увеличению роста на несколько сантиметров, увеличению молочных желез, появлению волосяного покрова на лобке, иногда появлению месячных.
К деторождению такие женщины не способны. Неизвестно, какого эффекта можно было бы достигнуть при своевременной диагностике этого заболевания и назначении необходимых гормональных препаратов.
Однако синдром Шерешевского — Тернера описан и у мужчин, у которых наблюдается нормальный кариотип XY, но у них Y-хромосома не активна. Правда, эти сообщения единичны.
Источник