Синдром веста по мкб 10
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
Названия
Название: Синдром Веста.
Синдром Веста
Описание
Синдром Веста. Серийные спастические сокращения в отдельных мышечных группах или генерализованного характера, протекающие на фоне задержки нейропсихического развития и сопровождающиеся гипсаритмическим ЭЭГ — паттерном. Манифестирует в возрасте до 4-х лет, преимущественно на 1-ом году жизни. В большинстве случаев имеет симптоматический характер. Диагностика синдрома основана на клинических данных и результатах ЭЭГ. Для выявления основной патологии необходимы КТ или МРТ, ПЭТ головного мозга, консультация генетика, нейрохирурга. Лечение возможно противоэпилептическими препаратами, стероидами (АКТГ, преднизолон), вигабатрином. По показаниям решается вопрос о хирургическом лечении (каллозотомия, удаление патологического очага).
Дополнительные факты
Синдром Веста носит название по имени врача, наблюдавшего его проявления у своего ребенка и впервые описавшего его в 1841 г. В связи с манифестацией синдрома в раннем возрасте и протеканием судорог по типу серии отдельных спазмов, пароксизмы, характеризующие синдром Веста, получили название инфантильные спазмы. Первоначально заболевание относили к генерализованной эпилепсии. В 1952 г. Был изучен специфический гипсаритмический ЭЭГ-паттерн, патогномоничный для этой формы эпилепсии и характеризующийся медленноволновой асинхронной активностью с беспорядочными спайками высокой амплитуды. В 1964 г. Специалистами в области неврологии синдром Веста был выделен в качестве отдельной нозологии.
Внедрение в неврологическую практику нейровизуализации позволило определить наличие у пациентов очаговых поражений вещества мозга. Это заставило неврологов пересмотреть свои взгляды на синдром Веста как на генерализованную эпилепсию и отнести его в ряд эпилептических энцефалопатий. В 1984 г. Был выявлена эволюция эпилептической формы энцефалопатии от ее раннего варианта в синдром Веста, а с течением времени в синдром Леннокса-Гасто.
В настоящее время синдром Веста занимает около 2% от всех случаев эпилепсии у детей и примерно четверть младенческой эпилепсии. Распространенность составляет, по различным источникам, от 2 до 4,5 случаев на 10 тыс. Новорожденных. Несколько чаще заболевают мальчики (60%). 90% случаев манифестации синдрома приходится на 1-й год жизни, с пиком в возрасте от 4 до 6 мес. Как правило, к возрасту 3-х лет мышечные спазмы проходят или трансформируются в иные формы эпилепсии.
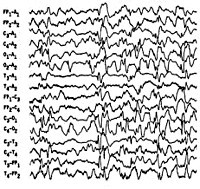
Синдром Веста
Причины
В подавляющем большинстве случаев синдром Веста носит симптоматический характер. Он может возникать вследствие перенесенных внутриутробных инфекций (цитомегалии, герпетической инфекции), постнатального энцефалита, гипоксии плода, преждевременных родов, внутричерепной родовой травмы, асфиксии новорожденного, постнатальной ишемии вследствие позднего пережатия пуповины. Синдром Веста может являться следствием аномалий строения головного мозга: септальной дисплазии, гемимегалоэнцефалии, агенезии мозолистого тела и пр. В ряде случаев инфантильные спазмы выступают симптомом факоматозов (синдрома недержания пигмента, туберозного склероза, нейрофиброматоза), точечных генных мутаций или хромосомных аберраций (в т. Синдрома Дауна). В литературе упоминаются случаи фенилкетонурии с инфантильными спазмами.
В 9-15% синдром Веста является идиопатическим или криптогенным, т. Е. Его первопричина не установлена или не очевидна. Зачастую при этом прослеживается наличие случаев фибрильных судорог или эпиприступов в семейном анамнезе больного ребенка, т. Е. Имеет место наследственная предрасположенность. Ряд исследователей указывают, что фактором, провоцирующим синдром Веста, может выступать вакцинация, в частности введение АКДС. Это может быть связано с совпадением сроков вакцинации и возраста типичного дебюта синдрома. Однако достоверные данные, подтверждающие провоцирующую роль вакцин, пока не получены.
Патогенетические механизмы возникновения инфантильных спазмов являются предметом изучения. Существует несколько гипотез. Одна из них связывает синдром Веста с расстройством функционирования серотонинергических нейронов. Действительно, у пациентов наблюдается понижение уровня серотонина и его метаболитов. Но пока неизвестно, является оно первичным или вторичным. Обсуждалась также иммунологическая теория, связывающая синдром Веста с увеличением количества активированных В-клеток. Положительный лечебный эффект АКТГ лег в основу гипотезы о сбоях в системе «мозг-надпочечники». Отдельные исследователи предполагают, что в основе синдрома лежит избыточное количество (гиперэкспрессия) возбуждающих синапсов и проводящих коллатералей, формирующих повышенную возбудимость коры. Асинхронность ЭЭГ-паттерна они связывают с физиологичным для этого возрастного периода недостатком миелина. По мере созревания мозга происходит уменьшение его возбудимости и нарастание миелинизации, что объясняет дальнейшее исчезновение пароксизмов или их трансформацию в синдром Леннокса-Гасто.
Симптомы
Как правило, симптом Веста дебютирует на первом году жизни. В отдельных случаях его манифестация происходит в более старшем возрасте, однако не позже 4-х лет. Основу клиники составляют серийные мышечные спазмы и нарушение психомоторного развития. Первые пароксизмы зачастую появляются на фоне уже существующей задержки психомоторного развития (ЗПР), но в 1/3 случаев возникают у первично здоровых детей. Отклонения в нейропсихологическом развитии наиболее часто проявляются снижением и выпадением хватательного рефлекса, аксональной гипотонией. Возможно отсутствие слежения глазами за предметами и расстройство фиксации взора, что является прогностически неблагоприятным критерием.
Мышечные спазмы носят внезапный симметричный и кратковременный характер. Типична их серийность, при этом интервал между следующими друг за другом спазмами длится не менее 1 минуты. Обычно наблюдается возрастание интенсивности спазмов в начале пароксизма и ее спад в конце. Число спазмов, происходящих за сутки, варьирует от единиц до сотен. Наиболее часто возникновение инфантильных спазмов происходит в период засыпания или сразу после сна. Провоцировать пароксизм способны резкие громкие звуки и тактильная стимуляция.
Семиотика пароксизмов, которыми сопровождается синдром Веста, зависит от того, какая мышечная группа сокращается — экстензорная (разгибательная) или флексорная (сгибательная). По этому признаку спазмы классифицируют на экстензорные, флексорные и смешанные. Чаще всего наблюдаются смешанные спазмы, затем сгибательные, наиболее редко — разгибательные. В большинстве случаев у одного ребенка наблюдаются спазмы нескольких видов и то, какой именно спазм будет преобладать, зависит от положения тела в момент начала пароксизма.
Может иметь место генерализованное сокращение всех мышечных групп. Но более часто наблюдаются локальные спазмы. Так, судороги в сгибателях шеи сопровождаются кивками головой, спазмы в мускулатуре плечевого пояса напоминают пожимание плечами. Типичным является пароксизм по типу «складного ножа», обусловленный сокращением мышц сгибателей живота. При этом тело как бы складывается пополам. Инфантильные спазмы верхних конечностей проявляются отведением и приведением рук к туловищу; со стороны кажется, что ребенок сам себя обнимает. Сочетание подобных спазмов с пароксизмом по типу «складного ножа» ассоциируется с принятым на Востоке приветствием «салаам», поэтому было названо «салаамовой атакой». У детей, которые умеют ходить, спазмы могут протекать по типу дроп-атак — неожиданных падений с сохранением сознания.
Судороги. Тонические судороги.
Диагностика
Синдром Веста диагностируется по основной триаде признаков: приступы кластерных мышечных спазмов, задержка психомоторного развития и гипсаритмический ЭЭГ-паттерн. Имеют значение возраст манифестации спазмов и их связь со сном. Трудности диагностики возникают при позднем дебюте синдрома. В ходе диагностики ребенок консультируется педиатром, детским неврологом, эпилептологом, генетиком.
Дифференциальная диагностика
Дифференцировать синдром Веста следует с доброкачественным младенческим миоклонусом, доброкачественной роландической эпилепсией, младенческой миоклонической эпилепсией, синдромом Сандифера (наклон головы по типу кривошеи, гастроэзофагальный рефлюкс, эпизоды опистотонуса, которые могут быть приняты за спазмы).
Интериктальная (межприступная) ЭЭГ характеризуется наличием дезорганизованной беспорядочной, динамично изменяющейся спайк-волновой активности, как в период бодрствования, так и во сне. Проведение полисомнографии позволяет выявить отсутствие спайк-активности в период глубоких стадий сна. Гипсаритмия регистрируется в 66% случаев, обычно на ранних стадиях. Позже наблюдается некоторая организация хаотичного ЭЭГ-паттерна, а в возрасте 2-4 лет его переход в комплексы «острая-медленная волна». Наиболее частый иктальный ЭЭГ-паттерн (т. Е. ЭЭГ-ритм в период спазмов) — это генерализованные медленноволновые комплексы высокой амплитуды с последующим угнетением активности не менее 1 сек. При регистрации на ЭЭГ фокальных изменений следует думать об очаговом характере поражения головного мозга или наличии аномалий его строения.
КТ головного мозга у имеющих синдром Веста детей может выявлять диффузные либо очаговые изменения церебральных структур, но может быть в пределах нормы. В диагностике локальных поражений более чувствительным методом является МРТ головного мозга. Для выявления участков гипометаболизма мозговых тканей в некоторых случаях возможно проведение ПЭТ головного мозга.
Лечение
Синдром Веста считался резистентным к проводимой терапии вплоть до открытия в 1958 г. Влияния на приступы препаратов АКТГ. Терапия АКТГ и преднизолоном приводит к значительному улучшению или полному прекращению инфантильных спазмов, что сопровождается исчезновением гипсаритмического ЭЭГ-паттерна. До сих пор среди неврологов нет однозначных решений касательно доз и длительности стероидной терапии. Исследования показали, что в 90% случаев терапевтический успех достигался при применении больших дозировок АКТГ. Сроки терапии могут варьировать в пределах 2-6 недель.
Новый этап в лечении инфантильных спазмов начался в 1990-1992 гг. После обнаружения положительного терапевтического эффекта вигабатрина. Однако преимущество лечения вигабатрином пока доказано лишь для больных туберозным склерозом. В остальных случаях исследования показали большую эффективность стероидов. С другой стороны стероидная терапия имеет худшую, в сравнении с вигабатрином, переносимость и более высокий процент рецидивов.
Из антиконвульсантов эффективность показана лишь у нитразепама и вальпроевой кислоты. У отдельных пациентов описан лечебный эффект больших доз витамина В6, который отмечался в первые недели терапии. При инфантильных спазмах, резистентных к проводимой терапии, с подтвержденным на томографии наличием патологического очага показана консультация нейрохирурга для решения вопроса о резекции очага. Если подобная операция невозможна, то при наличии дроп-атак проводится тотальная каллозотомия (пересечение мозолистого тела).
Прогноз
Обычно к 3-летнему возрасту наблюдается регресс и исчезновение инфантильных спазмов. Но примерно в 55-60% случаев они трансформируются в другую форму эпилепсии, чаще всего в синдром Леннокса-Гасто. Фармакорезистентность часто констатируется при инфантильных спазмах, сопровождающих синдром Дауна. Даже при успешном купировании пароксизмов синдром Веста имеет неудовлетворительный прогноз в плане психомоторного развития ребенка. Возможны когнитивные и поведенческие нарушения, ДЦП, аутизм, трудности в обучении. Остаточный психомоторный дефицит не наблюдается только в 5-12% случаев. ЗПР отмечается у 70-78% детей, двигательные расстройства — у 50%. Серьезный прогноз имеет синдром Веста, обусловленный аномалиями или дегенеративными изменениями головного мозга. При этом летальность может достигать 25%.
Более благоприятный прогноз имеют криптогенный и идиопатический синдром Веста при отсутствии ЗПР до появления спазмов. В этой группе больных остаточный интеллектуальный или неврологический дефицит отсутствует у 37-44% детей. Неблагоприятно отражается на прогнозе болезни откладывание начала лечения. Прогностическая оценка затрудняется тем, что отдаленные последствия также зависят от основной патологии, на фоне которой возникает симптоматический синдром Веста.
Источник
Утратил силу — Архив
![]()
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Архив — Клинические протоколы МЗ РК — 2007 (Приказ №764)
Категории МКБ:
Генерализованная идиопатическая эпилепсия и эпилептические синдромы (G40.3)
Общая информация
Краткое описание
Генерализованная эпилепсия (ГЭ) – хроническое заболевание головного мозга, характеризующиеся повторными приступами с нарушением двигательных, чувствительных, вегетативных, мыслительных или психических функций, возникающими вследствие чрезмерных нейронных разрядов в обоих полушариях головного мозга.
ГЭ – является единым заболеванием, представляющим отдельные формы с электро-клиническими особенностями, подходом к лечению и прогнозом.
Код протокола: H-P-003 «Генерализованная эпилепсия у детей, острый период»
Для стационаров педиатрического профиля
Код (коды) по МКБ-10:
G40.3 Генерализованная идиопатическая эпилепсия и эпилептические синдромы
G40.4 Другие виды генерализованной эпилепсии и эпилептических синдромов
G40.5 Особые эпилептические синдромы
G40.6 Припадки grand mal неуточненные (с малыми припадками (petit mal) или без них
G40.7 Малые припадки (petit mal) неуточненные, без припадков grand mal
G40.8 Другие уточненные формы эпилепсии G40.9 Эпилепсия неуточненная
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники. Стандарты лечения
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Классификация
Согласно Международной классификации 1989 г. (Международная лига борьбы с эпилепсией) в основу генерализованной эпилепсии положена генерализованность эпилептической активности.
Внутри ГЭ выделяют формы : идиопатические, симптоматические и криптогенные.
Генерализованные виды эпилепсии и синдромы:
1. Идиопатические (с возраст-зависимым дебютом). МКБ-10: G40.3:
— доброкачественные семейные неонатальные судороги;
— доброкачественные идиопатические неонатальные судороги;
— доброкачественная миоклоническая эпилепсия раннего детского возраста;
— детская абсансная эпилепсия (МКБ-10: G40.3);
— ювенильная абсансная эпилепсия;
— ювенильная миоклоническая эпилепсия;
— эпилепсия с приступами пробуждения;
— другие виды идиопатической генерализованной эпилепсии (МКБ-10: G40.4);
— эпилепсия с приступами, провоцируемыми специфическими факторами.
2. Криптогенные и (или) симптоматические (с возраст-зависимым дебютом) — МКБ-10: G40.5:
— синдром Веста (инфантильные спазмы);
— синдром Леннокса-Гасто;
— эпилепсия с миоклонически-астатическими приступами;
— эпилепсия с миоклоническими абсансами.
3. Симптоматические.
3.1 Неспецифической этиологии:
— ранняя миоклоническая энцефалопатия;
— ранняя инфантильная эпилептическая энцефалопатия с комплексами «вспышка-угнетение» на ЭЭГ;
— другие виды симптоматической генерализованной эпилепсии.
3.2 Специфические синдромы.
Диагностика
Диагностические критерии
Жалобы и анамнез
Особый акцент при сборе анамнеза:
— наследственность;
— на наличие в анамнезе неонатальных приступов, судорог при повышении температуры (являются факторами риска развития эпилепсии);
— токсические, ишемические, гипоксические, травматические и инфекционные поражения мозга, включая внутриутробный период (могут быть причинами данного заболевания).
Физикальное обследование:
— наличие судорог;
— характер приступов;
— семейная предрасположенность;
— возраст дебюта;
— длительность приступа.
Лабораторные исследования
Количество лейкоцитов и тромбоцитов определяют для исключения фолиево-дефицитной анемии и связанных с этим вторичных изменений костного мозга, что клинически проявляется снижением уровня лейкоцитов и тромбоцитов;
Снижение удельного веса мочи может свидетельствовать о появлении почечной недостаточности, что требует уточнения дозировки препаратов и тактики лечения.
Инструментальные исследования: данные ЭЭГ.
Показания для консультации специалистов: в зависимости от сопутствующей патологии.
Дифференциальный диагноз: нет.
Перечень основных диагностических мероприятий:
1. Эхоэнцефалография.
2. Общий анализ крови.
3. Общий анализ мочи.
Перечень дополнительных диагностических мероприятий:
1. Компьютерная томография головного мозга.
2. Ядерно-магнитно-резонансная томография головного мозга.
3. Консультация детского офтальмолога.
4. Консультация инфекциониста.
5. Консультация нейрохирурга.
6. Анализ ликвора.
7. Биохимический анализ крови.
Лечение
Первый врач, заставший эпилептический припадок, должен подробно его описать, включая признаки, которые предшествовали припадку и возникали после его окончания.
Больных необходимо направлять на полное неврологическое обследование для подтверждения диагноза и выяснения этиологии.
Лечение эпилепсии начинают только после установления точного диагноза. По мнению большинства специалистов, лечение эпилепсии следует начинать после повторного приступа.
Цели лечения:
— уменьшение частоты приступов;
— достижение ремиссии.
Немедикаментозное лечение: необходим полноценный ночной сон.
Медикаментозное лечение
Лечение эпилепсии должно осуществляться в зависимости от формы эпилепсии, а затем от характера приступов – с базового препарата для данной формы эпилепсии. Стартовая доза составляет примерно 1/4 от средней терапевтической. При хорошей переносимости препарата дозировка увеличивается примерно до 3/4 от средней терапевтической дозы в течение 2-3 недель.
При отсутствии или недостаточном эффекте доза повышается до средней терапевтической.
При отсутствии эффекта от терапевтической дозы в течение 1 месяца необходимо дальнейшее постепенное увеличение дозы до получения выраженного положительного эффекта или появления побочных эффектов.
При отсутствии терапевтического эффекта и появлении признаков интоксикации, препарат постепенно заменяется на другой.
При получении выраженного терапевтического эффекта и наличии побочных эффектов, необходимо оценить характер и степень выраженности последних, затем решить вопрос о продолжении лечения или замене препарата.
Замена барбитуратов и бензодиазепинов должна производиться постепенно в течение 2-4-х недель и более ввиду наличия выраженного синдрома отмены. Замена других антиэпилептических препаратов (АЭП) может быть осуществлена более быстро – за 1-2 недели. Оценка эффективности препарата может быть произведена лишь не ранее 1 месяца с момента начала его приема.
Противоэпилептические препараты, применяемые при генерализованных приступах судорог и ГЭ
Эпилептические приступы | Противоэпилептические препараты | ||
1-го выбора | 2-го выбора | 3-го выбора | |
Тонико-клонические | Вальпроаты | Дифенин Фенобарбитал Ламотриджин | |
Тонические | Вальпроаты | Дифенин Ламотриджин | |
Клонические | Вальпроаты | Фенобарбитал | |
Миоклонические | Вальпроаты | Ламотриджин Суксимиды Фенобарбитал | Клоназепам |
Атонические | Вальпроаты | Клобазам | |
Абсансы Типичные Атипичные Миоклонические | Вальпроаты Суксимиды Вальпроаты Ламотриджин Вальпроаты | Клоназепам Клобазам Клоназепам Клобазам Клоназепам | Кетогенная диета |
Отдельные формы эпилептических синдромов и эпилепсий | |||
Неонатальная миоклоническая энцефалопатия | Вальпроаты Карбамазепины | Фенобарбитал Кортикотропин | |
Инфантильная эпилептическая энцефалопатия | Вальпроаты Фенобарбитал | Кортикотропин | |
Осложненные фебрильные судороги | Фенобарбитал | Вальпроаты | |
Синдром Веста | Вальпроаты | Кортикотропин Нитразепам | Большие дозы пиридоксина Ламотриджин |
Синдром Леннокса- Гасто | Вальпроаты | Ламотриджин Иммуноглобулины | Кетогенная диета |
Синдром Леннокса- Гасто с тоническими приступами | Вальпроаты Топирамат Ламотриджин Фелбамат | Карбамазепины Сукцинимиды Бензодиазепины Гидантоиды | Кортикостероидные гормоны Иммуноглобулины Тиреотропин — релизинг гормон |
Миоклоническая астатическая эпилепсия | Вальпроаты | Клобазам Кортикотропин | Кетогенная диета |
Абсансная детская | Суксимиды | Вальпроаты | Клоназепам |
Абсансная детская сочетающаяся с генерализованными тонико-клоническими приступами | Вальпроаты | Дифенин Ламотриджин | Ацетазоламид (диакарб) |
Абсансная подростковая | Вальпроаты | Вальпроаты в сочетании с суксимидами | |
Миоклоническая ювенильная доброкачественная | Вальпроаты | Ламотриджин | Дифенин |
Эпилепсия пробуждения с генерализованными тонико-клоническими приступами | Вальпроаты Фенобарбитал | Ламотриджин | |
Средние суточные дозы АЭП ( мг/кг/сут.): фенобарбитал 3-5; гексамидин 20; дифенин 5-8; суксимиды (этосуксимид 15-30); клоназепам 0,1; вальпроаты 30-80; ламотриджин 2-5; клобазам 0,05-0,3-1,0; карбамазепины 5-15-30; ацетозоламид 5-10-20.
Перечень основных медикаментов:
1. *Вальпроевая кислота 150 мг, 300 мг, 500 мг табл.
2. Клобазам 500 мг,1000 мг табл.
3. Гексамидин 200 табл.
4. Этосуксимид 150-300 мг табл.
5. *Клоназепам 25 мг, 100 мг табл.
6. Карбамазепины 50-150-300 мг табл.
7. *Ацетозоламид 50-100-200 мг табл.
8. *Ламотриджин 25 мг, 50 мг табл.
Перечень дополнительных медикаментов:
1. *Дифенин 80 мг табл.
2. *Фенобарбитал 50 мг, 100 мг табл.
Дальнейшее ведение: диспансерное наблюдение.
Индикаторы эффективности лечения:
— урежение приступов;
— контроль за судорогами.
* – препараты, входящие в список основных (жизненно важных) лекарственных средств
Госпитализация
Показания для госпитализации:
— учащение приступов;
— резистентность к лечению;
— статусное течение;
— уточнение диагноза и формы эпилепсии.
Информация
Источники и литература
- Протоколы диагностики и лечения заболеваний МЗ РК (Приказ №764 от 28.12.2007)
- 1. Hopkins A., Appleton R. Epilepsy: Oxford University Press.1996.
2. Международная Классификация болезней 10 пересмотра;
3. Международная лига борьбы с эпилепсией (ILAE).Epilepsia 1989 vol. 30-P.389-399.
4. К.Ю.Мухин, А.С.Петрухин «Идиопатические эпилепсии: диагностика, тактика,
лечение».М.,2000 г.
5. Дигностика и лечение эпилепсий у детей .Под редакцией П.А.Темина,
М.Ю.Никаноровой, 1997 г.
6. Современные представления о детской эпилептической энцефалопатии с диффузными медленными пик-волнами (синдром Леннокса-Гасто).К.Ю. Мухин, А.С. Петрухин, Н.Б. Калашникова. Учебно-метод. Пособие. РГМУ, Москва, 2002 г.
7. Progress in Epileptic Disorders «Cognitive Dysfunction in Children with Temporal Lobe
Epilepsy». France, 2005.
8. Aicardi J. Epilepsy in children.-Lippincott- Raven, 1996.-Р.44-66.
9. Marson AG, Williamson PR, Hutton JL, Clough HE, Chadwick DW, on behalf of the epilepsy monotherapy trialists. Carbamazepine versus valproate monotherapy for epilepsy. In: The Cochrane Library, Issue 3, 2000;
10. Tudur Smith C, Marson AG, Williamson PR. Phenotoin versus valproate monotherapy for partial onset seizures and generalized onset tonic-clonic seizures. In: The Cochrane Library, Issue 4, 2001;
11. Доказательная медицина. Ежегодный справочник. Часть 2. Москва, Медиа Сфера, 2003. с 833-836.
12. First Seizure Trial Grroup (FIRST Group). Randomized clinical trial on the efficacy of
antiepilepic drugs in reducing the risk of relapse after a first unprovoked tonic clonic seizure. Neurology 1993;43: 478-483;
13. Medical Research Council Antiepileptic Drug Withdrawal Study Group. Randomised study of antiepilepic drug withdrawal in patients in remission. Lancet 1991; 337: 1175-1180.
14. Клинические рекомендации для практикующих врачей, основанные на доказательной медицине, 2-е издание. ГЭОТАР-МЕД, 2002, С. 933-935.
15. Never rugs for epilepsy in children. National Institute for Clinical Excellence. Technology Appraisal 79. April 2004. https://www.clinicalevidence.com.
16. Brodie MJ. Lamotrigine monoterapy: an overview. In: Loiseau P (ed). Lamictal – a brighter future. Royal Sosiety of Medicine Hress Ltd, London, 1996, pp 43-50.
17. O’Brien G et al. Lamotrigine in add-on terapy in treatment-resistant epilepsy in
mentallyhandicapped patients: an interim analisis. Epilepsia 1996, in press.
18. Karseski S., Morrell M., Carpenter D. The Expert Consensus Guideline Series: Treatment of Epilepsy. Epilepsia Epilepsy Behav. 2001; 2:A1-A50.
19. Hosking G et al. Lamotrigine in children with severe developmental abnormalities in a pediatric population with refractory seizures. Epilepsia 1993; 34 (Suppl): 42
20. Mattson RH. Efficacy and adverse effects of establiched and new antiepileptic drugs.
Epilepsia 1995; 36 (suppl 2): 513-526.
21. Калинин В.В., Железнова Е.В., Рогачева Т.А., Соколова Л.В., Полянский Д.А.,
Земляная А.А., Назметдинова Д.М. Применение препарата Магне В6 для лечения
тревожно-депрессивных состояний у больных эпилепсией. Журнал неврологии и
психиатрии 2004; 8: 51-55
22. Barry J., Lembke A., Huynh N. Affective disorders in epilepsy. In: Psychiatric issues in
epilepsy. A practical guide to diagnosis and treatment. A. Ettinger, A. Kanner (Eds.).
Philadelphia 2001; 45-71.
23. Blumer D., Montouris G., Hermann B. Psychiatric morbidity in seizure patients on a
neurodiagnostic monitoring unit. J. Neuropsychiat Clin Neurosci 1995; 7:445-446.
24. Edeh J., Toone B., Corney R. Epilepsy, psychiatric morbidity, and social dysfunction in
general practice. Comparison between hospital clinic patients and clinic non-attenders.
Neuropsychiat Neuropsychol Behav Neurol 1990; 3: 180-192.
25. Robertson M., Trimble M., Depressive illness in patients with epilepsy: a review. Epilepsia 1983; 24: Supple 2:109-116.
26. Schmitz B., Depressive disorders in epilepsy. In: Seizure, affective disorders and
anticonvusant drugs. M. Trimble, B. Schmitz (Eds.). UK 2002; 19-34.
- 1. Hopkins A., Appleton R. Epilepsy: Oxford University Press.1996.
Информация
Список разработчиков:
д.м.н., проф. Лепесова М.М., заведующая кафедрой детской неврологии АГИУВ
Прикреплённые файлы
Внимание!
Если вы не являетесь медицинским специалистом:
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта», не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта» являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.
Источник