Синдром ушера что это такое

Синдром Ушера (иногда синдром Ашера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) — сравнительно редкое генетическое заболевание, вызываемое мутацией одного из 10 генов, приводящее к врождённой нейросенсорной тугоухости и прогрессирующей потере зрения (пигментная дегенерация). Одна из основных причин слепоглухоты. В настоящее время неизлечим. Наследуется по аутосомно-рецессивному принципу.
Синдром Ушера — наиболее распространённая болезнь, приводящая к слепоглухоте[1][2]. В США с синдромом Ушера рождаются четверо из 100 тысяч новорожденных[3]. В Великобритании, по состоянию на 2010 год, проживали 132 тысячи слепоглухих — примерно 0,2 % населения[4].
Фоторецепторные клетки обычно дегенерируют от периферии к центру сетчатки, включая макулу (жёлтое пятно). Эта дегенерация сначала проявляется как куриная слепота (ослабление зрения при слабом освещении). Периферическое зрение постепенно теряется, ограничивая поле зрения до туннельного, постепенно приводя к полной слепоте[5].
Существует несколько[6][7][8][9][10] классификаций синдрома Ушера, но наиболее распространённой является классификация по степени глухоты[11].
- I тип, встречается с частотой 3-6 на 100 тысяч человек, сопровождается врождённой глубокой тугоухостью или полной глухотой и нарушением вестибулярных функций, раннее начало пигментного ретинита. Связан с мутациями в генах CDH23, MYO7A, PCDH15, USH1C, USH1G. Более распространён среди евреев-ашкеназов.
- II тип, сопровождается тугоухостью, не ухудшающейся со временем, вестибулярные функции не нарушены. Пигментная дегенерация сетчатки начинается с возраста 10-20 лет. Связан с мутациями генов USH2A, GPR98, DFNB31.
- III тип, прогрессирующее ухудшение слуха и зрения, примерно в половине случаев сопровождается вестибулярными нарушениями. Чаще встречается у финнов. Связан с мутациями гена CLRN1.
Литература[править | править код]
- C. H. Usher. On the inheritance of retinitis pigmentosa with notes of cases. Royal London Ophthalmological Hospital Report, 1914; 19: 130—336.
- Stiefel S. H., Lewis R. A. The Madness of Usher’s: Coping With Vision and Hearing Loss/Usher Syndrome Type II (англ.). — Business of Living Publications, 1991. — ISBN 978-1-879518-06-3.
- Duncan E., Prickett H. T. Usher’s Syndrome: What It Is, How to Cope, and How to Help (англ.). — Charles C. Thomas, 1988. — ISBN 978-0-398-05481-6.
- Vernon M. Answers to your questions about Usher’s syndrome (retinitis pigmentosa with hearing loss) (англ.). — Foundation Fighting Blindness, 1986.
- Vernon M. Usher’s syndrome: Deafness and progressive blindness : clinical cases, prevention, theory and literature survey (англ.). — Pergamon Press (англ.)русск., 1969.
Примечания[править | править код]
- ↑ Vernon M. Usher’s syndrome—deafness and progressive blindness. Clinical cases, prevention, theory and literature survey. (англ.) // Journal of chronic diseases. — 1969. — Vol. 22, no. 3. — P. 133—151. — doi:10.1016/0021-9681(69)90055-1. — PMID 4897966.
- ↑ Балашова Л.М., Гонтаренко Ю.Э. и др. Положение людей с выраженными нарушениями слуха и зрения (слепоглухих) в Российской Федерации. Отчёт по результатам исследования (pdf) (недоступная ссылка). Институт политики детства и прикладной социальной работы (2015). Дата обращения 1 апреля 2017. Архивировано 13 июля 2017 года.
- ↑ American deaf-blind population (англ.). LibGuides (July 2010). Дата обращения 1 апреля 2017.
- ↑ Estimating the Number of People with Co‐Occurring Vision and Hearing Impairments in the UK (англ.) (pdf) (недоступная ссылка). Centre for Disability Research (April 2010). Дата обращения 1 апреля 2017. Архивировано 1 апреля 2017 года.
- ↑ Williams D. S. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. (англ.) // Vision research. — 2008. — Vol. 48, no. 3. — P. 433—441. — doi:10.1016/j.visres.2007.08.015. — PMID 17936325.
- ↑ Gorlin R. J., Tilsner T. J., Feinstein S., Duvall A. J. 3rd. Usher’s syndrome type III. (англ.) // Archives of otolaryngology (Chicago, Ill. : 1960). — 1979. — Vol. 105, no. 6. — P. 353—354. — doi:10.1001/archotol.1979.00790180051011. — PMID 454290.
- ↑ Merin S., Auerbach E. Retinitis pigmentosa. (англ.) // Survey of ophthalmology. — 1976. — Vol. 20, no. 5. — P. 303—346. — doi:10.1016/S0039-6257(96)90001-6. — PMID 817406.
- ↑ Fishman G. A., Kumar A., Joseph M. E., Torok N., Anderson R. J. Usher’s syndrome. Ophthalmic and neuro-otologic findings suggesting genetic heterogeneity. (англ.) // Archives of ophthalmology (Chicago, Ill. : 1960). — 1983. — Vol. 101, no. 9. — P. 1367—1374. — doi:10.1001/archopht.1983.01040020369005. — PMID 6604514.
- ↑ Sankila E. M., Pakarinen L., Kääriäinen H., Aittomäki K., Karjalainen S., Sistonen P., de la Chapelle A. Assignment of an Usher syndrome type III (USH3) gene to chromosome 3q. (англ.) // Human molecular genetics. — 1995. — Vol. 4, no. 1. — P. 93—98. — doi:10.1093/hmg/4.1.93. — PMID 7711740.
- ↑ HALLGREN B. Retinitis pigmentosa combined with congenital deafness; with vestibulo-cerebellar ataxia and mental abnormality in a proportion of cases: A clinical and genetico-statistical study. (англ.) // Acta psychiatrica Scandinavica. Supplementum. — 1959. — Vol. 34, no. 138. — P. 1—101. — doi:10.1111/j.1600-0447.1959.tb08605.x. — PMID 14399116.
- ↑ Smith R. J., Berlin C. I., Hejtmancik J. F., Keats B. J., Kimberling W. J., Lewis R. A., Möller C. G., Pelias M. Z., Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. (англ.) // American journal of medical genetics. — 1994. — Vol. 50, no. 1. — P. 32—38. — doi:10.1002/ajmg.1320500107. — PMID 8160750.
Ссылки[править | править код]
- Ирина Кравцова. Не лечится, помочь нельзя.. Что такое синдром Ушера и как живут слепоглухие в России.. Meduza (31 марта 2017). Дата обращения 1 апреля 2017.
- Что такое синдром Ушера? // по материалам Конференции по проблемам слепоглухоты и синдрома Ушера, март 1999, Москва.
- СИНДРОМ УШЕРА // «ОБЪЕДИНЕНИЕ СЛАБОСЛЫШАЩИХ»
Источник
Синдром Ушера – редкое генетическое заболевание, характеризующееся глухотой из-за нарушения способности внутреннего уха и слуховых нервов передавать сенсорный (звуковой) сигнал в мозг (сенсорная потеря слуха).
Сопровождается пигментной ретинитой, расстройством, поражающим сетчатку и вызывающим прогрессирующую потерю зрения. Определено три клинических типа синдрома. Их отличают возраст появления симптомов и тяжесть. Usher наследуется как аутосомно-рецессивный генетический признак.
Введение
Впервые описан в 1858 году Альбрехтом фон Графе, назван в честь Чарльза Ушера, шотландского офтальмолога, идентифицировавшего наследственный, рецессивный характер наследования заболевания.
Признаки
Характеризуется глухотой из-за нарушения способности внутреннего уха и слуховых нервов передавать сенсорный (звуковой) вход в мозг (сенсорную потерю слуха). Также, аномальное скопление цветного (пигментного) материала на сетчатке (пигментный ретинит или РП).
РП вызывает дегенерацию сетчатки, приводящую к постепенной потере зрения и полной слепоте.
Сенсинологическая глухота нерва бывает глубокой, легкой, прогрессивной. Потеря зрения, вызванная пигментным ретинитом, начинается в детстве или позже в течение жизни. Часто впервые проявляется как плохое видение ночью или при низкой освещенности («ночная слепота»).
Исследования показывают, что четкое центральное зрение поддерживается в течение многих лет, даже когда боковое (периферическое) уменьшается.
Типы болезни
- Синдром Ушера типа 1 характеризуется глубокой потерей слуха при рождении (врожденная глухота), так и проблемами с балансом. Во многих случаях пострадавшие дети не умеют ходить до 18 месяцев или позже.
Проблемы со зрением начинаются в раннем подростковом возрасте, около десяти лет или немного раньше.
- Тип 2 характеризуется умеренной, иногда тяжелой потерей слуха в обоих ушах при рождении. Потеря слуха ухудшается с течением времени. Начало ночной слепоты происходит во время позднего подросткового возраста или начала двадцати лет.
Потери периферического зрения продолжаются, но центральное обычно сохраняется во взрослую жизнь. Визуальные проблемы имеют тенденцию прогрессировать медленнее, чем при типе 1.
- Тип 3 характеризуется более поздними нарушениями слуха, вариабельным балансом (вестибулярной) дисфункцией и РП, появляющейся между вторым и четвертым десятилетиями жизни.
Вестибулярные нарушения встречаются примерно у 50% детей с синдромом Ушера типа 3.
| 1 | 2 | 3 | |
| Слух | Глубокая потеря слуха или глухота при рождении. | От умеренной до тяжелой потери слуха при рождении. | Прогрессивная потеря в детстве или раннем подростковом возрасте. |
| Зрение | Снижение ночного видения к возрасту 10, прогрессирование к серьезной потери зрения на середине жизни. | Снижение ночного видения в подростковом возрасте, прогрессирование к серьезным потерям зрения на середине жизни. | Изменяется степень тяжести и возраста начала; проблемы ночного видения часто начинаются в подростковом возрасте и прогрессируют до тяжелой потери зрения в середине жизни. |
| Баланс (вестибулярная функция) | Проблемы с балансом с рождения. | Нормальный баланс. | От нормального до почти нормального равновесия в детстве; вероятность более поздних проблем. |

Почему возникает
Синдром ушера вызван мутациями определенных генов. До сих пор он ассоциировался с мутациями по меньшей мере в десяти:
- Тип 1: MYO7A (USH1B), USH1C, CDH23, PCDH15 (USH1F), SANS (USH1G), возможно, CIB2;
- 2 тип: USH2A, ADGRV1 (ранее называемый VLGR1), WHRN (DFNB31);
- Вариант 3: USH3A (CLRN1), HARS.
Эти гены предоставляют инструкции для нормального слуха, зрения, равновесия. Некоторые из них помогают специализированным клеткам, называемым волосковыми, передавать звук от внутреннего уха до мозга, ощущать свет и цвет сетчатке глаза. Функция некоторых белков, связанных с расстройством неизвестна.
Некоторые люди не имеют мутаций ни в одном из этих генов, поэтому, существуют другие, связанные с синдромом, которые еще не были идентифицированы.

Все типы синдрома Ушера наследуются как аутосомно-рецессивные черты. Большинство генетических заболеваний определяются статусом двух копий гена, полученных от отца и матери.
Когда наследуется один ген заболевания и один нормальный, то человек становится носителем без проявления симптомов.
- Риск того, что оба родителя-носителя передадут измененный ген детям – 25%.
- Риск иметь ребенка, который является носителем, таким как родители, составляет 50%.
- Шанс на получение ребенком нормальных генов от обоих родителей – 25%.
Риск одинаковый для мужчин и женщин.
Родители, близкие родственники имеют более высокий шанс, чем неродственные связи. Ненормальный ген, увеличивает риск иметь детей с рецессивным генетическим расстройством.
Затронутые популяции
Синдром Usher затрагивает три – десять из 100 000 человек во всем мире. Среди людей еврейской национальности Израиля, Берлина, Германии было обнаружено более высокое, чем среднее число пострадавших.
Часто поражаются Французские канадцы Луизианы, Аргентинцы испанского происхождения, нигерийские африканцы.
Тип 3, самая редкая форма в большинстве популяций, составляет около 40% пациентов Финляндии. Это самое распространенное генетическое заболевание, связанное с потерями зрения, слуха. Примерно 10 процентов всех случаев умеренной и глубокой глухоты у детей случается из-за типа 1 и 2.

Связанные нарушения
Симптомы следующих расстройств бывают сходны, сравнения полезны для постановки дифференциального диагноза:
Синдром Alström
Наследственное расстройство, характеризующееся дегенерацией сетчатки с нистагмом и потерей центрального зрения. Заболевание связано с ожирением в детстве. Сенсинологическая глухота и сахарный диабет развиваются после десятилетнего возраста.
Краснуха (немецкая корь)
Острое вирусное расстройство, вызывающее беспокойство при болезни на первых трех месяцев беременности, поскольку может вызвать отклонения плода. Эти аномалии включают нарушения слуха, зрения, пороки развития у ребенка.
Пигментный ретинит (RP)
Содержит большую группу наследственных нарушений зрения, вызывающих прогрессирующую дегенерацию сетчатки.
Периферическое (боковое) зрение постепенно уменьшается и теряется в большинстве случаев.
Центральное зрение обычно остается сохранно до позднего возраста. Некоторые формы RP связаны с глухотой, ожирением, заболеваниями почек, другими проблемами здоровья, включая поражения центральной нервной системы, нарушения обмена веществ, хромосомные аномалии
Диагностика
Синдром Ушера диагностируется при помощи обследования слуха, координации движения и зрения. При слуховом (аудиологическом) осмотре измеряется частота и громкость звуков, которые слышит человек.

Электроретинограмма измеряет электрический ответ на светочувствительных клетках сетчатки глаза. Осмотр сетчатки проводится для наблюдения за структурами задней части глаза.
Вестибулярную (балансовую) функцию оценивают с помощью тестов на разные части системы. Генетическое тестирование доступно для большинства генов, связанных с расстройством.

Лечение
Лечение синдрома Ушера направлено на конкретные симптомы. Оно требует скоординированных усилий группы медицинских специалистов, таких как педиатры, терапевты, отоларингологи, аудиологи, офтальмологи и другие специалисты здравоохранения.
Следует оценивать чувствительность органов дыхания. Возможности коммуникации изучаются как можно раньше, чтобы обеспечить ребенку прочную языковую базу. Слуховые аппараты или кохлеарные имплантаты принесут пользу большинству младенцев и детей с синдромом Ушера.
Язык жестов можно изучить как вариант связи. Люди, которые теряют зрение, изучают тактильные знаки. Раннее вмешательство важно для того, чтобы дети с синдромом Ушера достигли своего потенциала. Для детей с сенсоневральной глухотой или глухонемой требуются дополнительные медицинские, социальные, профессиональные услуги.

В настоящее время нет лечения расстройства, хотя исследователи работают над генетической и другой терапией для восстановления потери зрения, слуха.
Терапия витаминами
Исследования показали, для людей с типичным синдромом Ушера и типом 2, прием определенной суточной дозы витамина А замедляет дегенерацию сетчатки. Эксперты рекомендуют, чтобы взрослые пациенты с общими формами расстройства принимали витамин А пальмитат 15 000 МЕ ежедневно под наблюдением офтальмологов. Соблюдали регулярную сбалансированную диету и избегали приема витамина Е высоких доз.
Поскольку долгосрочная добавка витамина А высокой дозой (превышающая 25 000 МЕ) вызывает побочные эффекты, такие как болезни печени. Поэтому пациентам необходимо регулярно контролироваться врачами при приеме таких добавок.
Лица с пигментным ретинитом в сочетании с синдромом Ушера могут рассчитывать на помощь офтальмологов по коррекции слабого зрения. Другое лечение является симптоматическим и поддерживающим.
Генетическая консультация рекомендуется для пострадавших лиц и их семей.
Понравилась статья? Поделись с друзьями:
Источник
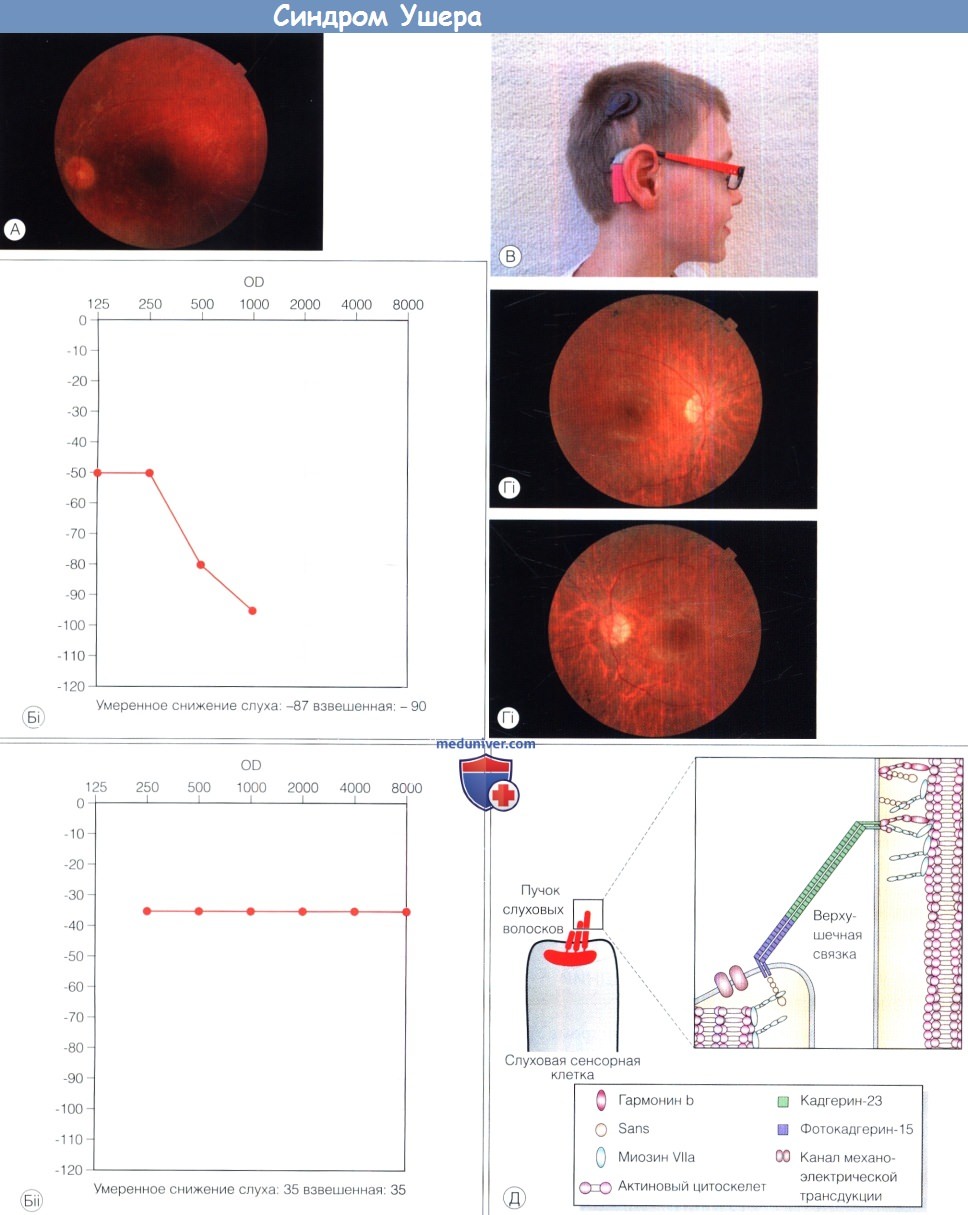
Синдром Ушера (Usher’s syndrome, USH) — глухой слепнущий ребенокСиндром Ушера (Usher’s syndrome, USH) — комбинация сенсоневральной глухоты с прогрессирующей дегенерацией сетчатки. Синдромом Ушера страдает один из каждых десяти глухих детей (5% всех случаев врожденной глухоты). Это наиболее часто встречающийся наследственный синдром, сопровождающийся глухотой и наиболее часто встречающийся синдром среди слепоглухонемых. Синдром подразделяется на три группы: синдром Ушера 1 типа (USH1), 2 типа (USH2) и 3 типа (USH3). Синдром Ушера 1 и 2 типов обычно диагностируется в раннем или старшем детском возрасте, соответственно, но может диагностироваться и позже, поскольку ухудшение зрения у глухого ребенка может оставаться незамеченным. Вестибулярная дисфункция связана с классической картиной синдрома Ушера 1 типа. Каждый тип синдрома Ушера наследуется по аутосомно-рецессивному механизму, но наблюдается генетическая гетерогенность этих заболеваний. Для каждого типа идентифицировано несколько генов. С помощью молекулярных исследований выявлено клиническое частичное совпадение классических первого и второго типов синдрома Ушера. Некоторые гены Ушера могут вызывать развитие изолированных глухоты или пигментного ретинита (USH2A). Таким детям настоятельно рекомендуется применение кохлеарных имплантов (особенно при USH1). Лечение дегенерации сетчатки все еще не разработано. а) Синдром Ушера 1 типа: наиболее тяжелая форма. Синдром Ушера 1 типа проявляется у детей тяжелой или полной сенсоневральной глухотой, ребенок поздно начинает садиться и ходить вследствие нарушения вестибулярной функции. В раннем детстве развивается дегенерация сетчатки. Уже в возрасте 2-3 лет изменения ЭРГ подтверждают диагноз дегенерации сетчатки, хотя глазное дно выглядит нормальным. Известно пять генов, мутации которых вызывают развитие заболевания, их частота варьирует в зависимости от исследуемой популяции: MY07A (считается основным геном, вызывающим синдром Ушера 1 типа, его мутации выявляются в половине случаев; также мутации этого гена вызывают несиндромальную глухоту), USH1C, CDH23, PCDH15 и USH1G. Хотя заболевание всегда протекает тяжело, его течение может различаться в зависимости от типа мутации. б) Синдром Ушера 2 типа: более позднее развитие дегенерации сетчатки у глухого ребенка. Врожденная сенсоневральная стабильная глухота от легкой до тяжелой степени тяжести, выраженные нарушения в области высоких частот и нормальная вестибулярная функция — основные признаки синдрома Ушера 2 типа. Дегенерация сетчатки развивается позже, чем при синдроме Ушера 1 типа, обычно в пубертатном периоде или позже. Синдром Ушера 2 типа и синдром Ушера 3 типа обычно не проявляются в течение первого десятилетия жизни и первоначально не вызывают развития изменений глазного дна. У многих пациентов сохраняется хорошая острота зрения, несмотря на сужение полей зрения. На ЭРГ на ранних стадиях изменения могут отсутствовать; однако изменения ЭРГ могут выявляться у детей и до развития клинической картины. Во втором десятилетии жизни болезнь проявляется ночной слепотой и утратой периферического зрения и необратимо прогрессирует. Идентифицировано три гена: USH2A (при изолированном пигментном ретините также выявлен мутантный ушерин), GPR98 и DFNB31. в) Синдром Ушера 3 типа: более поздний дебют, течение вариабельно. Возраст дебюта пигментного ретинита при синдроме Ушера 3 типа вариабелен, изменения сетчатки часто выявляются в возрасте старше двадцати лет, но могут быть ошибочно приняты за проявления двух других типов заболевания. Сенсорная глухота развивается уже постлингвально (в отличие от прелингвального развития глухоты при синдроме Ушера 1 и 2 типов), речь у таких пациентов развита нормально, но болезнь прогрессирует и приводит к полной слепоте. Вестибулярная дисфункция различной тяжести развивается у половины пациентов. Ген синдрома Ушера 3 типа особенно часто выявлялся у евреев-ашкенази и финнов. У пациентов с синдромом Ушера 1 и 2 типа также обнаружен мутантный USH3. г) Патогенез синдрома Ушера. В изучении патогенеза этого заболевания достигнут прогресс, установлено, что поражаются слуховые волоски внутреннего уха и фоторецепторные клетки сетчатки. Продукты гена USH1 формируют сеть, интерактом Ушера, который играет ключевую роль в раннем развитии пучков слуховых волосков. Они также являются ключевым компонентом механизма механоэлектрической трансдукции, необходимого для реализации функции слуха. В сетчатке интерактом Ушера действует на уровне цилиарной/пери-цилиарной зоны, в области соединения соединительного волоска с фоторецепторной клеткой. Эти структуры играю важную роль в транспорте протеинов между наружными и внутренними сегментами. Дефекты протеинов USH вызывают рано дебютирующую дисфункцию как внутреннего уха, так и сетчатки.
— Вернуться в содержание раздела «офтальмология» на сайте Оглавление темы «Наследственная патология сетчатки.»:
|
Источник