Синдром умственной отсталости с ломкой х хромосомой

Генетические особенности синдрома ломкой Х-хромосомы
Среди группы наследственных болезней есть два заболевания, относящихся к самым частым причинам интеллектуальной недостаточности. Самая известная и наиболее распространённая патология – синдром Дауна, связанный с наличием лишней 21-ой хромосомы в геноме человека. В этой статье мы расскажем о втором по распространенности наследственном заболевании, которое приводит к умственной отсталости, а также может сопровождаться другими клиническими проявлениями.
Синдром ломкой X-хромосомы или синдром Мартина-Белл является результатом нарушения в гене FMR1 (fragile X mental retardation-1), который расположен на Х-хромосоме и играет важную роль в появлении и развитии нервных связей, обучении и запоминании. Частота этого синдрома среди мальчиков составляет 1:4000.
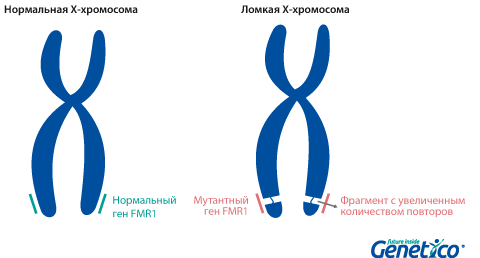
Так называемая «ломкость» X-хромосомы проявляется в том, что хромосома выглядит нетипично при специальном окрашивании, как будто один кусок отделился, хотя физически она остается цельной. Генетическая основа этого явления заключается в увеличении числа тринуклеотидных повторов CGG в гене FMR1, расположенном на X-хромосоме.
У здоровых людей число повторов в этом гене колеблется от 5 до 54. Если повторов больше 200, то наработка белка с гена FMR1 нарушается, что приводит к развитию синдрома Мартина-Белл и клиническому проявлению заболевания. Премутационное состояние — это количество повторов CGG от 55 до 200. В таком состоянии заболевание у людей в типичной форме не проявляется, но чем больше повторов в этом гене у носителя, тем больше вероятность того, что у ее или его детей количество повторов будет больше 200 и заболевание разовьется. В случае носительства премутации при формировании половых клеток количество повторов может увеличиваться, поэтому если у родителя количество повторов от 55 до 200, то высока вероятность рождения ребенка с мутантным геном FMR1 и синдромом Мартина-Белл. При этом носительство премутационного состояния будущим папой и мамой неравнозначно по вероятности возникновения мутантного аллеля у их детей: если носитель – мама, то вероятность значительного увеличения числа повторов гораздо выше. Количество повторов от 45 до 54 является промежуточной формой, которая не имеет никакого влияния на здоровье человека, но может приводить к проблемам у будущих поколений, как и в случае премутационного состояния гена.
Важно учитывать, что наследование и развитие заболевания зависит от пола, так как ген FMR1 находится на Х-хромосоме. У мужчин только одна Х-хромосома, которую они получают от матери. Поэтому, в случае, если эта одна хромосома оказалась «ломкой», у них проявляется заболевание. У женщин две Х-хромосомы, однако активно работает только одна из них. Поэтому наличие одной Х-хромосомы с мутантным геном FMR1 может не проявляться клинически, в случае инактивации именно «ломкой» хромосомы, или приводить к развитию заболевания в 30-50% случаев. Мужчина с ломкой Х-хромосомой может передать её всем дочерям, но ни одному из сыновей. Женщина с мутантной хромосомой имеет шансы передать её как сыновьям, так и дочерям с равной вероятностью.
Премутационное состояние гена влияет как на судьбу потомков носителя такого гена, так и непосредственно на его здоровье:
Развитие первичной недостаточности яичников (FXPOI) (снижение овариального резерва и наступление менопаузы до 40 лет). Мутация FMR1 является причиной преждевременного истощения яичников у 5% женщин с этим диагнозом. Среди носительниц премутации примерно у четверти развивается это состояние. Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя бедный ответ яичников на стимуляцию влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.
Тремор/атаксия, ассоциированные с ломкой Х-хромосомой (FXTAS). Это состояние чаще развивается у мужчин: при носительстве премутации мужчиной проявляется в 33% случаев, а при носительстве премутации женщиной – лишь в 5-10%. Синдром FXTAS начинает проявляться в пожилом возрасте. Наблюдается тремор, шаткая походка, может страдать речь.
Метод диагностики, используемый в лаборатории Genetico, основан на использовании полимеразной цепной реакции с особым набором праймеров, позволяющих не только детектировать нормальное, премутационное и мутационное состояния, но и точно определить количество повторов в случаях, когда их меньше 200. Такая диагностика позволяет выявить синдром ломкой X-хромосомы на молекулярном уровне, а также оценить вероятность рождения ребенка с этим синдромом и возможность развития у пациента расстройств, связанных с увеличенным количеством повторов в гене FMR1. Такая диагностика также позволяет детектировать наличие AGG повторов среди повторов CGG. Полагают, что участки AGG, прерывающие длинную последовательность из CGG повторов, придают ДНК устойчивость и снижают риск увеличения количества повторов в следующем поколении.
Генетический тест, определяющий количество повторов в гене FMR1, рекомендуется пройти в первую очередь женщинам с синдромом преждевременного истощения яичников или с выявленной неслучайной инактивацией Х-хромосомы (косвенный признак), семьям, в которых есть сыновья с интеллектуальной недостаточностью. Также анализ состояния гена FMR1 необходим:
1) женщинам с репродуктивными проблемами или нарушениями фертильности, связанными с повышенным уровнем фолликулостимулирующего гормона (ФСГ)
2) пациентам с интеллектуальной недостаточностью и их родственникам
3) тем, у кого в семье были случаи синдрома ломкой Х-хромосомы или умственной отсталости без точного диагноза
4) женщинам, у родственников которых наблюдались нарушения, связанные с премутационным состоянием FMR1
5) пациентам с поздно проявившимся тремором и мозжечковой атаксией (нарушения согласованности работы мышц из-за поражения систем мозга, управляющих движением мышц).
В случае обнаружения бессимптомного носительства мутации в гене FMR1 у женщины может быть рекомендовано использование донорских ооцитов или проведение преимплантационной генетической диагностики (ПГД) с целью исключить возможность проявления синдрома у ребенка. Также важно правильно оценивать риск рождения больного ребенка в случае премутационного состояния гена FMR1 у будущих родителей. В таком случае по результатам теста рекомендуется консультация врача-генетика.
Автор: Очир Мигяев
Стажер лаборатории Genetico
Источник
Синоним: синдром Мартина-Белла. Ранее эту болезнь называли синдромом олигофрении с маркерной Х-хромосомой. Синдром Мартина-Белла — одна из наиболее часто встречающихся (после болезни Дауна) форм умственной отсталости. Популяционная частота заболевания составляет 1:2000-1:5000 всех живорождённых. Больных мальчиков в 2-3 раза больше, чем девочек, мальчики болеют тяжелее.
Внешность больных неспецифична, но определённое диагностическое значение имеют удлинённое лицо, высокий выступающий лоб, макро- и долихоцефалия, гипоплазированная средняя часть лица (особенно в сравнении с выступающими и часто увеличенными щеками), выступающий подбородок. Нёбо обычно дугообразное, губы толстые, нижняя губа часто вывернута. Отмечается также увеличенный размер оттопыренных ушных раковин. Обращают на себя внимание большие кисти и стопы. Одно из типичных проявлений синдрома — макроорхидизм. При рождении у детей яички нормальных размеров, но с наступлением пубертатного периода существенно увеличиваются. Увеличение размеров гонад происходит за счёт избыточного роста соединительной ткани и накопления жидкости, но половая активность таких больных минимальна, способны к половой жизни единицы.
Могут отмечаться повышенная растяжимость кожных покровов, слабость связочного аппарата суставов (что приводит к самопроизвольными вывихам, обычно пальцев кисти), плоскостопие; у многих больных формируется пролапс митрального клапана. Все эти симптомы обусловлены врождённой дисплазией соединительной ткани.
Умственная отсталость, типичная для больных с синдромом Мартина-Белла, как правило, оценивается как умеренная или выраженная, хотя у 10-15% больных обнаруживается глубокая олигофрения и ещё у такого же процента больных — «мягкая» умственная отсталость. Большинство пациентов социально адаптированы, выполняют несложную физическую работу. Психологи оценивают их как контактных и доброжелательных. Лишь в тяжёлых случаях таких больных необходимо помещать в специализированные интернаты. Неврологические симптомы включают в себя мышечную гипотонию и в отдельных случаях судороги.
Пороки развития и нарушения других органов сравнительно редки: расщелины нёба, нистагм, страбизм, птоз, катаракта, кривошея в сочетании со сколиозом, кифоз, дефект межпредсердной перегородки.
Помимо определённого комплекса клинических аномалий, синдром Мартина-Белла сопровождается характерной цитогенетической картиной: ломкостью в дистальной части длинного плеча Х-хромосомы (в зоне Xq), что внешне напоминает «спутник» длинного плеча. Эта ломкость выявляется лишь при культивировании лимфоцитов в условиях дефицита фолиевой кислоты, поэтому для обнаружения ломкости нужно либо использовать культуральные среды, лишённые фолиевой кислоты, либо вводить в культуральную среду антагонисты фолиевой кислоты, но даже и при этих условиях ломкость Х-хромосомы выявляется не во всех клетках (до 60%).
Длительное время считалось, что синдром наследуется по Х-сцепленному рецессивному типу. Однако в родословных отмечались случаи тяжело протекающей болезни у женщин и легко протекающей у мужчин. У женщин-гетерозигот отмечали некоторое снижение интеллекта, чаще оцениваемое как пограничная умственная отсталость. У некоторых из них в небольшом проценте клеток обнаруживали ломкую Х-хромосому. Таким образом, наследование синдрома Мартина-Белла не укладывалось в строгие рамки Х-сцепленного рецессивного наследования. Более того, в родословных отмечалась антиципация. Этиология этого заболевания была выяснена с помощью методов молекулярно-генетического анализа. Была обнаружена экспансия нестабильных тринуклеотидных повторов (CGG) в 5′-нетранслируемой области FMR-I гена (fragile menial retardation). В норме в этом гене число повторов варьирует от 6 до 42. Хромосомы, в которых имеется 50-200 повторов, считаются «премутацией». В следующем поколении число повторов может увеличиться (экспансия) до 1000 и более, что и обусловит выраженную клиническую картину, зависящую именно от числа повторов. Соответственно если женщина унаследовала большое число повторов, то она будет больной. Обнаружены ещё два гена, мутации в которых обусловливают такую же клиническую картину, но эти гены встречаются редко.
Диагноз ставят на основании клинической картины и по результатам кли-нико-генеалогического и цитогенетического исследований. Наиболее точный метод — молекулярно-генетическая диагностика.
В настоящее время возможна пренатальная диагностика данного синдрома. Этиотропной терапии пока не существует.
Источник
Синдром умственной отсталости с ломкой Х-хромосомой. (синдром Мартина-Белла).
Ранее эту болезнь называли синдромом олигофрении с маркерной Х-хромосомой. Синдром Мартина-Белла — одна из наиболее часто встречающихся (после болезни Дауна) форм умственной отсталости. Популяционная частота заболевания составляет 1:2000-1:5000 всех живорождённых. Больных мальчиков в 2-3 раза больше, чем девочек, мальчики болеют тяжелее.
Внешность больных неспецифична, но определённое диагностическое значение имеют удлинённое лицо, высокий выступающий лоб, макро- и долихоцефалия, гипоплазированная средняя часть лица (особенно в сравнении с выступающими и часто увеличенными щеками), выступающий подбородок. Нёбо обычно дугообразное, губы толстые, нижняя губа часто вывернута. Отмечается также увеличенный размер оттопыренных ушных раковин. Обращают на себя внимание большие кисти и стопы. Одно из типичных проявлений синдрома — макроорхидизм. При рождении у детей яички нормальных размеров, но с наступлением пубертатного периода существенно увеличиваются. Увеличение размеров гонад происходит за счёт избыточного роста соединительной ткани и накопления жидкости, но половая активность таких больных минимальна, способны к половой жизни единицы.
Могут отмечаться повышенная растяжимость кожных покровов, слабость связочного аппарата суставов (что приводит к самопроизвольными вывихам, обычно пальцев кисти), плоскостопие; у многих больных формируется пролапс митрального клапана. Все эти симптомы обусловлены врождённой дисплазией соединительной ткани.
Умственная отсталость, типичная для больных с синдромом Мартина-Белла, как правило, оценивается как умеренная или выраженная, хотя у 10-15% больных обнаруживается глубокая олигофрения и ещё у такого же процента больных — «мягкая» умственная отсталость. Большинство пациентов социально адаптированы, выполняют несложную физическую работу. Психологи оценивают их как контактных и доброжелательных. Лишь в тяжёлых случаях таких больных необходимо помещать в специализированные интернаты. Неврологические симптомы включают в себя мышечную гипотонию и в отдельных случаях судороги.
Пороки развития и нарушения других органов сравнительно редки: расщелины нёба, нистагм, страбизм, птоз, катаракта, кривошея в сочетании со сколиозом, кифоз, дефект межпредсердной перегородки.
Помимо определённого комплекса клинических аномалий, синдром Мартина-Белла сопровождается характерной цитогенетической картиной: ломкостью в дистальной части длинного плеча Х-хромосомы (в зоне Xq), что внешне напоминает «спутник» длинного плеча. Эта ломкость выявляется лишь при культивировании лимфоцитов в условиях дефицита фолиевой кислоты, поэтому для обнаружения ломкости нужно либо использовать культуральные среды, лишённые фолиевой кислоты, либо вводить в культуральную среду антагонисты фолиевой кислоты, но даже и при этих условиях ломкость Х-хромосомы выявляется не во всех клетках (до 60%).
Длительное время считалось, что синдром наследуется по Х-сцепленному рецессивному типу. Однако в родословных отмечались случаи тяжело протекающей болезни у женщин и легко протекающей у мужчин. У женщин-гетерозигот отмечали некоторое снижение интеллекта, чаще оцениваемое как пограничная умственная отсталость. У некоторых из них в небольшом проценте клеток обнаруживали ломкую Х-хромосому. Таким образом, наследование синдрома Мартина-Белла не укладывалось в строгие рамки Х-сцепленного рецессивного наследования. Более того, в родословных отмечалась антиципация. Этиология этого заболевания была выяснена с помощью методов молекулярно-генетического анализа. Была обнаружена экспансия нестабильных тринуклеотидных повторов (CGG) в 5′-нетранслируемой области FMR-I гена (fragile menial retardation). В норме в этом гене число повторов варьирует от 6 до 42. Хромосомы, в которых имеется 50-200 повторов, считаются «премутацией». В следующем поколении число повторов может увеличиться (экспансия) до 1000 и более, что и обусловит выраженную клиническую картину, зависящую именно от числа повторов. Соответственно если женщина унаследовала большое число повторов, то она будет больной. Обнаружены ещё два гена, мутации в которых обусловливают такую же клиническую картину, но эти гены встречаются редко.
Диагноз ставят на основании клинической картины и по результатам кли-нико-генеалогического и цитогенетического исследований. Наиболее точный метод — молекулярно-генетическая диагностика.
В настоящее время возможна пренатальная диагностика данного синдрома. Этиотропной терапии пока не существует
Источник

Синоним: синдром Мартина-Белла. Ранееэту болезнь называли синдромом олигофрении с маркерной Х-хромосомой.Синдром Мартина-Белла — одна из наиболее часто встречающихся (послеболезни Дауна) форм умственной отсталости. Популяционная частотазаболевания составляет 1:2000-1:5000 всех живорождённых. Больныхмальчиков в 2-3 раза больше, чем девочек, мальчики болеют тяжелее.
Внешность больных неспецифична, но определённоедиагностическое значение имеют удлинённое лицо, высокий выступающийлоб, макро- и долихоцефалия, гипоплазированная средняя часть лица(особенно в сравнении с выступающими и часто увеличенными щеками),выступающий подбородок. Нёбо обычно дугообразное, губы толстые, нижняягуба часто вывернута. Отмечается также увеличенный размер оттопыренныхушных раковин. Обращают на себя внимание большие кисти и стопы. Одно изтипичных проявлений синдрома — макроорхидизм. При рождении у детейяички нормальных размеров, но с наступлением пубертатного периодасущественно увеличиваются. Увеличение размеров гонад происходит за счётизбыточного роста соединительной ткани и накопления жидкости, нополовая активность таких больных минимальна, способны к половой жизниединицы.
Могут отмечаться повышенная растяжимость кожныхпокровов, слабость связочного аппарата суставов (что приводит ксамопроизвольными вывихам, обычно пальцев кисти), плоскостопие; умногих больных формируется пролапс митрального клапана. Все этисимптомы обусловлены врождённой дисплазией соединительной ткани.
Умственная отсталость, типичная для больных ссиндромом Мартина-Белла, как правило, оценивается как умеренная иливыраженная, хотя у 10-15% больных обнаруживается глубокая олигофрения иещё у такого же процента больных — «мягкая» умственная отсталость.Большинство пациентов социально адаптированы, выполняют несложнуюфизическую работу. Психологи оценивают их как контактных идоброжелательных. Лишь в тяжёлых случаях таких больных необходимопомещать в специализированные интернаты. Неврологические симптомывключают в себя мышечную гипотонию и в отдельных случаях судороги.
Пороки развития и нарушения других органовсравнительно редки: расщелины нёба, нистагм, страбизм, птоз, катаракта,кривошея в сочетании со сколиозом, кифоз, дефект межпредсерднойперегородки.
Помимо определённого комплекса клинических аномалий, синдром Мартина-Белласопровождается характерной цитогенетической картиной: ломкостью вдистальной части длинного плеча Х-хромосомы (в зоне Xq), что внешненапоминает «спутник» длинного плеча. Эта ломкость выявляется лишь прикультивировании лимфоцитов в условиях дефицита фолиевой кислоты,поэтому для обнаружения ломкости нужно либо использовать культуральныесреды, лишённые фолиевой кислоты, либо вводить в культуральную средуантагонисты фолиевой кислоты, но даже и при этих условиях ломкостьХ-хромосомы выявляется не во всех клетках (до 60%).
Длительное время считалось, что синдром наследуетсяпо Х-сцепленному рецессивному типу. Однако в родословных отмечалисьслучаи тяжело протекающей болезни у женщин и легко протекающей умужчин. У женщин-гетерозигот отмечали некоторое снижение интеллекта,чаще оцениваемое как пограничная умственная отсталость. У некоторых изних в небольшом проценте клеток обнаруживали ломкую Х-хромосому. Такимобразом, наследование синдрома Мартина-Белла неукладывалось в строгие рамки Х-сцепленного рецессивного наследования.Более того, в родословных отмечалась антиципация. Этиология этогозаболевания была выяснена с помощью методов молекулярно-генетическогоанализа. Была обнаружена экспансия нестабильных тринуклеотидныхповторов (CGG) в 5′-нетранслируемой области FMR-I гена (fragile menialretardation). В норме в этом гене число повторов варьирует от 6 до 42.Хромосомы, в которых имеется 50-200 повторов, считаются «премутацией».В следующем поколении число повторов может увеличиться (экспансия) до1000 и более, что и обусловит выраженную клиническую картину, зависящуюименно от числа повторов. Соответственно если женщина унаследовалабольшое число повторов, то она будет больной. Обнаружены ещё два гена,мутации в которых обусловливают такую же клиническую картину, но этигены встречаются редко.
Диагноз ставят на основании клинической картины и порезультатам кли-нико-генеалогического и цитогенетического исследований.Наиболее точный метод — молекулярно-генетическая диагностика.
В настоящее время возможна пренатальная диагностика данного синдрома. Этиотропной терапии пока не существует.
Источник