Синдром тестикулярной феминизации мкб 10

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром тестикулярной феминизации.
Синдром тестикулярной феминизации
Описание
Синдром тестикулярной феминизации. Наследственное заболевание, характеризующееся развитием женских половых признаков при наличии мужского кариотипа (XY). Симптомы этой патологии обладают широким спектром выраженности – от фенотипически полноценной женщины до полноценного мужчины с целым рядом промежуточных вариантов, на чем построена классификация данного состояния. Диагностика синдрома тестикулярной феминизации производится на основании результатов гинекологического или урологического осмотра, ультразвуковых исследований органов малого таза, изучения кариотипа и молекулярно-генетических анализов. Специфического лечения этого заболевания не существует, для улучшения качества жизни больных применяются разнообразные хирургические вмешательства.
Дополнительные факты
Синдром тестикулярной феминизации – генетическое заболевание, поражающее лиц с мужским кариотипом и приводящее к развитию у них разнообразных по выраженности женских половых признаков вплоть до полной феминизации. Нарушения подобного типа регистрировались давно, сразу после открытий хромосомных основ пола и исследований кариотипа, но данное заболевание впервые описал американских гинеколог Джон Моррис в 1953 году. Он изучил известных на тот момент (более 80) пациентов с мужским псевдогермафродитизмом и выявил ряд семейных случаев, что позволило ему определить синдром тестикулярной феминизации как X-сцепленное рецессивное наследственное заболевание. В некоторых источниках эту патологию можно найти под названием синдрома Морриса. В настоящий момент установлено, что встречаемость синдрома тестикулярной феминизации составляет примерно 1 случай на 20-60 тысяч новорожденных мужского пола, однако частота носительства патологического гена среди женщин неизвестна. Данное заболевание является причиной почти 20% случаев мужского псевдогермафродитизма и обуславливает заметную долю от всех разновидностей первичной аменореи.
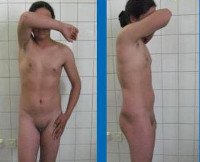
Синдром тестикулярной феминизации
Причины
Исследования в области современной генетики позволили выявить молекулярно-генетические механизмы синдрома тестикулярной феминизации – таковыми оказались мутации гена AR, локализованного на X-хромосоме. Продуктом экспрессии данного гена является белок-рецептор к тестостерону и его метаболитам (в основном, дигидротестостерону), наличие которого и обеспечивает реакцию организма на мужские половые гормоны. На сегодняшний день выявлено более 300 различных типов мутаций гена AR, приводящих к синдрому тестикулярной феминизации. Все они имеют рецессивный характер, поэтому женщины (по кариотипу), имеющие гомологичную Х-хромосому, могут выступать только в качестве носителя и передавать это заболевание своим сыновьям с вероятностью 50%.
Из-за нарушений в структуре гена AR кодируемый им белок-рецептор получается дефектным, в зависимости от типа мутаций его реакция на воздействие тестостерона и сходных с ним соединений изменяется. При наиболее тяжелом течении синдрома тестикулярной феминизации рецептор становится совсем неспособным взаимодействовать с мужскими половыми гормонами, поэтому клетки организма теряют к ним чувствительность, сохраняя ее к эстрогенам (в основном, к эстрадиолу). Это приводит к развитию организма полностью по женскому типу при наличии и функционировании яичек. Некоторые другие типы синдрома тестикулярной феминизации обусловлены сохранением чувствительности к тестостерону, но на крайне низком уровне, что становится причиной широкого спектра клинических проявлений. Активность клеток Сертоли, выделяющих тестостерон, при любой форме синдрома тестикулярной феминизации сохраняется и может быть даже несколько повышена.
Крайняя форма синдрома тестикулярной феминизации имеет следующий патогенез – еще на этапе эмбрионального развития из-за отсутствия влияния тестостерона по причине нечувствительности к нему организма происходит формирование женских половых органов. Образуется «слепое» влагалище, расщепленная мошонка становится большими половыми губами, зачатки фаллоса – клитором. На этапе полового созревания также полностью отсутствует реакция клеток на тестостерон, поэтому при синдроме тестикулярной феминизации (полный тип) ткани организма испытывают влияние только женских половых гормонов. Данное обстоятельство приводит к формированию выраженных вторичных половых признаков – пышной груди, «модельных» форм, распределению жировой клетчатки по женскому типу, тонкому голосу. Однако менструации и, тем более, возможность беременности в этом случае полностью исключены, так как у больных отсутствует матка и необходимые для менструального цикла гормональные факторы.
Классификация
Синдром тестикулярной феминизации характеризуется обширным спектром выраженности проявлений и разновидностей заболевания, для их систематизации была разработана специальная классификация данной патологии. В первую очередь, все формы этого состояния делятся на две группы – полные и неполные. Полная форма синдрома тестикулярной феминизации возникает при полном отсутствии чувствительности организма к тестостерону и характеризуется полноценным женским фенотипом. Обычно рождается здоровая девочка, не имеющая, на первый взгляд, каких-либо отклонений в развитии. С началом полового созревания такие больные нередко становятся весьма красивыми (с «модельной внешностью») из-за превалирующего влияния эстрогенов. Однако при этом у них отсутствует оволосение в области подмышечных впадин и на лобке, не наступает менархе – зачастую именно аменорея в возрасте 14-16 лет становится поводом для обращения к врачу, где в рамках гинекологического исследования и выявляется синдром тестикулярной феминизации.
Неполная форма синдрома тестикулярной феминизации характеризуется намного более разнообразной клинической картиной. Как правило, причиной этой формы патологии выступают дефекты рецепторов к тестостерону, не приводящие к полной потере чувствительности, а только к ее значительному снижению или нарушению. Согласно классификации, принятой в 1996 году, выделяют пять основных форм или степеней неполного синдрома тестикулярной феминизации:
Симптомы
1. 1. Я степень (мужской тип) характеризуется типично мужским фенотипом без каких — либо отклонений. В крайне редких случаях может наблюдаться высокий голос и признаки гинекомастии в подростковом возрасте. При этом практически всегда нарушены процессы сперматогенеза, поэтому у больных данным типом синдрома тестикулярной феминизации наблюдается мужское бесплодие.
2. 2. Я степень (преимущественно мужской тип) — данный вариант заболевания проявляется более выраженными нарушениями вирилизации и формирования половых органов, хотя фенотипически больные являются мужчинами. У них часто обнаруживается гипоспадия, возможно развитие микропениса или сочетание этих признаков. Пациенты с этим вариантом синдрома тестикулярной феминизации нередко имеют гинекомастию и неравномерное отложение подкожной жировой клетчатки.
3. 3. Я степень (амбивалентный тип) — характеризуется выраженным уменьшением полового члена, который становится похожим на клитор. Мошонка разделена настолько, что ее половины напоминают большие половые губы, часто наблюдается гипоспадия, крипторхизм. При этом типе синдрома тестикулярной феминизации также отмечается расширение таза, гинекомастия, относительно узкие плечи.
4. 4. Я степень (преимущественно женский тип) — при этой форме синдрома тестикулярной феминизации больные являются женщинами по своим фенотипическим признакам, однако их клитор часто гипертрофирован, урогенитальный синус формирует короткое слепое влагалище. Нередко при этом варианте заболевания наблюдается такое нарушение, как сращение половых губ.
5. 5. Я степень (женский тип) — больные этой формой синдрома тестикулярной феминизации фенотипически являются женщинами, практически никаких признаков вирилизации у них не обнаруживается за исключением несколько увеличенного размера клитора. В период полового созревания он может увеличиваться еще больше, достигая размеров микропениса.
Диагностика
Основными методами диагностики этого состояния являются гинекологический или урологический осмотр, ультразвуковое исследование, изучение наследственного анамнеза, молекулярно-генетический анализ и определение уровня половых гормонов. Раньше всего удается диагностировать неполные формы синдрома тестикулярной феминизации 2-5 степеней, так как нарушения в строении половых органов заметны уже при рождении ребенка. При осмотре врач-неонатолог может заподозрить наличие генетического заболевания и назначить дополнительные уточняющие исследования. Сочетание таких пороков развития с нормальным или даже повышенным уровнем тестостерона в крови и крипторхизмом говорит в пользу наличия синдрома тестикулярной феминизации.
Неполная форма заболевания 1 степени и полный тип патологии в большинстве случаев определяются намного позднее. Поводом для обращения мужчин (по своим фенотипическим признакам) с синдромом тестикулярной феминизации к специалистам часто служит подозрение на бесплодие. Анализ спермы при этом выявляет азооспермию, в анамнезе больного присутствует крипторхизм (нередко устраненный оперативным путем), паховые грыжи. Подтвердить наличие синдрома тестикулярной феминизации в этом случае возможно только при помощи генетической диагностики. Полная форма заболевания чаще всего диагностируется в 14-15 лет, когда девушки обращаются к специалисту из-за отсутствия менструаций. При гинекологическом осмотре у них определяется «слепое», закрытое в верхней трети влагалище, ультразвуковые исследования обнаруживают отсутствие матки и ее придатков. При этом могут выявляться семенники, находящие на различных этапах спуска в мошонку – располагающиеся в пределах брюшной полости, пахового канала, изредка в половых губах.
Концентрация тестостерона при синдроме тестикулярной феминизации соответствует уровню здорового мужчины или даже несколько превышает его. При этом количество эстрогенов не достигает нижней отметки нормы для девушек аналогичного возраста. Изучение наследственного анамнеза может обнаружить признаки Х-сцепленной передачи заболевания. Молекулярно-генетическая диагностика синдрома тестикулярной феминизации производится врачом-генетиком при помощи автоматического секвенирования последовательности гена AR или других методик. Возможно также выявление носительства патологической формы гена у здоровых женщин и пренатальная диагностика этого заболевания.
Лечение
Лечение синдрома тестикулярной феминизации полного типа часто ограничивается простым удалением семенников с последующей коррекцией гормонального фона (при необходимости). Это требуется для профилактики семиномы и других форм злокачественного перерождения тканей яичка. Такие больные, с детства воспитывающиеся как девочки, после постановки диагноза могут нуждаться в психологической помощи. Терапия синдрома тестикулярной феминизации неполного типа характеризуется большим количеством пластических операций для воссоздания привычного вида половых органов, груди, оптимального функционирования мочевыделительной системы. Больные с любой формой этого заболевания бесплодны, что также зачастую требует помощи психологов.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: E34.5
МКБ-10 / E00-E90 КЛАСС IV Болезни эндокринной системы, расстройства питания и нарушения обмена веществ / E20-E35 Нарушения других эндокринных желез / E34 Другие эндокринные нарушения
Определение и общие сведения[править]
Синонимы:
— синдром тестикулярной феминизации;
— синдром Морриса (полная форма синдрома резистентности к андрогенам);
— синдром Рейфенштейна (неполная форма синдрома резистентности к андрогенам).
Частота
1:20 400-1:99 000 новорожденных мальчиков. В России частота данного заболевания неизвестна.
Этиология и патогенез[править]
Синдром резистентности к андрогенам имеет Х-сцепленный рецессивный характер наследования, представляет собой полную или частичную нечувствительность тканей к мужским половым гормонам вследствие дефекта гена AR.
Клинические проявления[править]
1. Полная форма:
— правильное женское строение наружных гениталий, в некоторых случаях — недоразвитие клитора, больших и малых половых губ, слепо заканчивающееся влагалище и отсутствие матки;
— в период полового созревания — формирование феминной фигуры, развитие молочных желез (вплоть до макромастии);
— лобковое оволосение отсутствует или минимально развито.
2. Неполная форма:
— наружные половые органы могут иметь любую степень вирилизации — от клиторомегалии до практически правильно сформированных мужских гениталий;
— степень формирования производных Вольфовых протоков зависит от степени чувствительности к андрогенам;
— в период полового созревания развивается гинекомастии, выраженность которой не зависит от степени маскулинизации;
— снижение полового влечения у взрослых пациентов, олигоили азооспермия в спермограмме.
Синдром андрогенной резистентности: Диагностика[править]
Кариотип: 46XY.
— Полная форма. В раннем детском возрасте диагноз устанавливают после выявления у девочки яичек в содержимом паховых грыж, которые чаще бывают двусторонними. Среди пациентов с паховой грыжей и женским фенотипом распространенность андрогенной нечувствительности составляет 1-2%. У большей части пациентов диагноз ставят в пубертатный период при обращении по поводу первичной аменореи.
— Неполную форму клинически диагностируют при появлении гинекомастии в подростковом возрасте.
— В период полового созревания отмечают повышенную концентрацию ЛГ в сыворотке крови, уровень ФСГ нормальный или слегка повышенный, концентрация тестостерона повышена, уровень эстрогенов соответствует верхней мужской норме или несколько повышен.
— При молекулярно-генетическом исследовании выявляют мутации в гене AR.
Дифференциальный диагноз[править]
Синдром андрогенной резистентности: Лечение[править]
При адаптации в женском поле:
— хирургическое лечение при полной форме синдрома резистентности к андрогенам включает гонадэктомию;
— при неполной форме добавляют этап феминизирующей пластики и проведение интроитопластики в подростковом возрасте.
В 10-11 лет начинают заместительную терапию препаратами эстрогенов (этинилэстрадиол) — пероральными или трансдермальными формами. Для удлинения влагалища используют бескровный кольпопоэз, что позволяет большинству пациенток избежать вагинопластических операций.
Большинство детей с неполной формой синдрома резистентности к андрогенам воспитаны в мужском поле в связи с поздней постановкой диагноза. Хирургическая коррекция включает орхидопексию, исправление хорды и маскулинизирующую пластику для коррекции гипоспадии. Для увеличения размера полового члена используют местное применение тестостероновой или дигидротестостероновой мази.
В период полового созревания у пациентов развивается гинекомастия, которая требует хирургической коррекции.
Взрослым пациентам внутримышечно назначают высокие дозы препаратов тестостерона.
Профилактика[править]
Проведение пренатальной и предимплантационной диагностики в семьях, где есть пациенты с верифицированным диагнозом.
Прочее[править]
Осложнения
— Опухоли яичек у пациентов с синдромом резистентности к андрогенам встречают чаще, чем в здоровой популяции.
— Риск злокачественных опухолей зародышевых клеток с возрастом увеличивается: приблизительно от 3% в возрасте до 20 лет до 30% к 50 годам, поэтому гонадэктомию рекомендовано проводить до 18 лет.
Источники (ссылки)[править]
Детская эндокринология. Атлас [Электронный ресурс] / под ред. И. И. Дедова, В. А. Петерковой. — М. : ГЭОТАР-Медиа, 2016. — https://www.rosmedlib.ru/book/ISBN9785970436141.html
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Синдром нечувствительности к андрогенам, или синдром тестикулярной феминизации (300068, ген AR рецептора андрогенов [313700], Xq11–q12, À, рецессив]), выявляют у пациенток с мужским кариотипом (46,ХY), но женским фенотипом, половые органы сформированы по женскому типу. Частота тестикулярной феминизации — 1:65 000 генетических мужчин.
Этиология и патогенез • Синдром тестикулярной феминизации (синдром Морриса) — тип мужского псевдогермафродитизма: наличие наружных женских половых органов, недоразвитие репродуктивных органов, вторичных половых признаков и аменорея; образуются как андрогены, так и эстрогены, но рецепторы в значительной степени рефрактерны к андрогенам; у больных нет полового хроматина, они имеют нормальный мужской кариотип • Обычно у новорождённых мальчиков содержание тестостерона повышено в течение нескольких месяцев. Содержание тестостерона, превышающее нормальные показатели, при одновременном увеличении содержания ЛГ может указывать на резистентность к андрогенам либо на нарушение отрицательной обратной связи (ингибирование синтеза тестостерона). Это соотношение сохраняется и у взрослых.
Клиническая картина и диагностика
• При полной резистентности к андрогенам ребёнок с генотипом 46,XY (имеющий яички) внешне выглядит как девочка. Ключ к диагностике — обнаружение яичек в паховом канале. В подростковом возрасте таких детей расценивают как девочек с первичной аменореей. Поскольку яички во внутриутробном периоде синтезировали мюллеров ингибирующий фактор, влагалище представляет собой неглубокий и слепо оканчивающийся карман. Если яички не были удалены до пубертата из-за происходящего в них превращения тестостерона в эстроген, развиваются нормальные грудные железы.
• При частичной устойчивости к андрогенам больной с кариотипом XY имеет гениталии переходного типа. Диагноз установить трудно. Поскольку патология наследуется по X-сцепленному рецессивному типу, мать ребёнка должна быть носителем генного дефекта, а половина её детей должны быть либо девочками (XY-генотип, также носители), либо больными мальчиками. Таким образом, в семейном анамнезе надо искать указания на бесплодие или крипторхизм.
МКБ-10 • E34.5 Синдром андрогенной резистентности
Источник