Синдром сильвера рассела код по мкб 10

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Причины
- Симптомы
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Синдром Рассела-Сильвера.
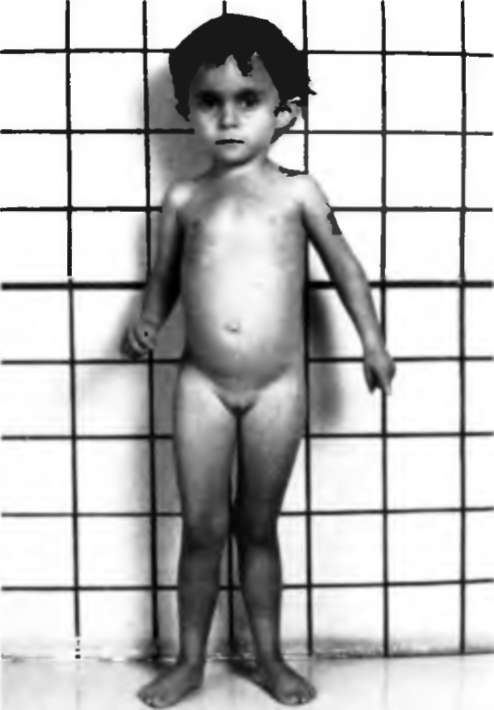
Синдром Рассела-Сильвера
Описание
Синдром Рассела-Сильвера врожденное заболевание, характеризующееся внутриутробной задержкой роста плода, поздним закрытием большого родничка, нарушением формирования скелета. Тип наследования неизвестен. Встречается в большинстве случаев спорадически, однако описаны и единичные родословные с данной патологией. Частота в популяции 1:30 000. Встречается в равной степени, как у мужчин, так и у женщин.
Причины
Причиной заболевания считаются генетические нарушения, приводящие к подобным порокам развития. Дети с данной патологией имеют малую длину и массу тела при рождении. Значительная задержка роста и костного созревания наблюдается и в дальнейшем.
Главной причиной преждевременного полового созревания при этом заболевании является избыток гонадотропных гормонов.
Симптомы
Дети рождаются небольшой длины (до 45 см) и с малой массой тела (1,5—2,5 кг). С годами отставание в росте сохраняется, в связи, с чем окончательный рост у женщин менее 150 см, у мужчин немногим выше 150 Масса тела у взрослых нормальная или даже избыточная. Часты аномалии мочевыделительной системы и наружных половых органов: крипторхизм, гипоспадия, гипоплазия полового члена, мошонки. Характерна асимметрия тела (лицо, туловище, длина ног). Лицо треугольной формы: псевдогидроцефалия, большой лоб и гипоплазия нижней челюсти, высокое нёбо, нередко с расщелиной, оттопыренные уши. Узкая грудная клетка, короткие руки, поясничный лордоз. Половое развитие опережает возраст ( в 30% случаев), начинается в 5-6 лет и обусловлено гонадотропной стимуляцией яичников. Интеллект сохранен в большинстве случаев.
Лечение
С 1985 г. Для лечения детей с соматотропной недостаточностью используются исключительно генно-инженерные препараты гормона роста человека.
В настоящее время в России прошли клиническую апробацию и разрешены к использованию следующие рекомбинантные препараты гормона роста человека: Нордитропин RНордиЛетR (Ново Нордиск, Дания); хуматроп (Лилли Франс, Франция); генотропин (Пфайзер Хелс АБ, Швеция); сайзен (Индустрия Фармасьютика Серано С. А. , Италия); растан (Фармстандарт, Россия).
При лечении гипофизарного нанизма у детей имеется четкая связь «доза–ростовой эффект», особенно выраженная в первый год лечения. Рекомендуемая стандартная доза СТГ при терапии классического дефицита СТГ – 0, 033 мг/кг/на инъекцию, ежедневно, подкожно, в вечернее время 20,00-22,00.
Критерием эффективности терапии является увеличение скорости роста от исходной в несколько раз. Она достигает в первый год лечения, по данным разных авторов, от 8 до 13 см в год. Максимальная скорость роста отмечается в первый год лечения, особенно в первые 3-6 мес, затем имеет место замедление скорости роста от первого ко второму году лечения (при сохранении скорости роста более 5-6 см в год).
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Причины
- Патогенез
- Симптомы
- Лечение
Другие названия и синонимы
Синдром гипотонии-ожирения.
Названия
Синдром Прадера — Вилли.
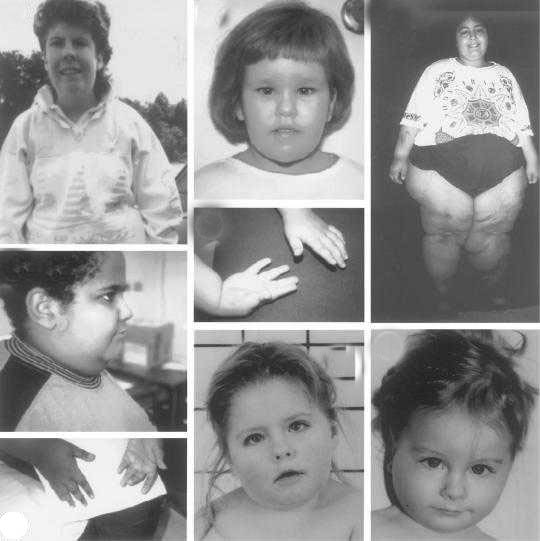
Проявления синдрома Прадера — Вилли
Синонимы диагноза
Синдром гипотонии-ожирения.
Описание
Синдром Прадера — Вилли — редкая генетическая аномалия. При синдроме Прадера — Вилли отсутствуют или не экспрессируются примерно 7 генов из 15-й хромосомы, унаследованной от отца.
Кариотип 46 XX или ХУ, 15q-11-13. Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г.
По данным регистра ассоциации больных с синдромом Прадера-Вилли, в США и Канаде на декабрь 1986 г. Насчитывалось 1595 больных. В последние годы удалось установить популяционную частоту патологии, составляющую 1 : 10 000 — 1 : 20 000.
Причины
Авторы, впервые описавшие синдром, высказывали предположение об аутосомно-рецессивном типе наследования заболевания. Затем появились сообщения о возможности аутосомно-доминантной передачи болезни. Подтверждением данных гипотез могли служить наблюдавшиеся семейные случаи патологии. Однако большинство описанных клинических наблюдений синдрома Прадера — Вилли носило спорадический характер.
Последующие исследования позволили установить у детей с синдромом Прадера — Вилли определенные хромосомные нарушения. Цитогенетический анализ показал, что хромосомные аномалии у больных были представлены либо транслокациями (t 15/15), либо мозаицизмом. В 1987 г. Появились первые сообщения о микроделеции хромосомы 15. Однако окончательная идентификация хромосомных изменений при синдроме Прадера — Вилли стала возможной только после внедрения в практику молекулярно-генетических методов исследования.
В настоящее время установлено, что развитие синдрома Прадера — Вилли связано с повреждением критического района хромосомы 15 (сегмента q11,2- q13). При этом оказалось, что повреждение этого же участка хромосомы 15 наблюдается и при другом заболевании — синдроме Ангельмана, клиническая картина которого существенно отличается от синдрома Прадера — Вилли и характеризуется ранним (в возрасте 6-12 мес) замедлением психомоторного развития, микроцефалией, нарушением речи (в 100% случаев), атаксией, неконтролируемым насильственным смехом, частыми эпилептиформными припадками, специфическим выражением лица.
Таким образом, несмотря на повреждение при синдромах Прадера — Вилли и Ангельмана одного и того же локуса хромосомы 15, клинические проявления обеих болезней резко противоположны.
Объяснение фенотипических различий получено лишь в последние годы. Оказалось, что развитие этих заболеваний связано с новыми генетическими явлениями — геномным импринтингом и унипарентальной дисомией.
Геномный импринтинг — новое явление, открытое благодаря успехам молекулярной генетики. Он означает различную экспрессию генетического материала (гомологичных аллелей) в хромосомах в зависимости от отцовского или материнского происхождения, т. Е. Свидетельствует о влиянии родителей на фенотип ребенка. До настоящего времени считалось, что вклад в проявляемость (экспрессию) генов отца и матери равноценен.
По сути геномный импринтинг — это половой и тканевозависимый сложный модификатор генной активности некоторых локусов хромосом в зависимости от их родительского происхождения. Проявления геномного импринтинга выявлены и при других заболеваниях — синдромах Сотоса, Беквита-Видемана, Сильвера-Рассела, муковисцидозе и других.
Унипарентальная (однородительская) дисомия — наследование обеих хромосом только от одного из родителей. В течение многих лет считалось, что такое наследование невозможно. Лишь с помощью молекулярно-генетических маркеров удалось доказать возможность однородительской дисомии. Природа унипарентальной дисомии окончательно не выяснена, однако установлено, что она обязана своим происхождением ряду генетических и биохимических нарушений.
Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить микроделецию или унипарентальную дисомию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы — прометафазный анализ, использование ДНК-маркеров определенных участков хромосомы 15 (исследование процессов метилирования) и.
На сегодняшний день синдромы Прадера — Вилли и Ангельмана служат общепринятой моделью для изучения новых в клинической генетике и сложных явлений — геномного импринтинга и унипарентальной дисомии.
Установлено, что синдром Прадера — Вилли может быть обусловлен двумя основными механизмами. Первый из них — микроделеция хромосомы 15 (15q11,2-q13), которая всегда отцовского происхождения. Второй — материнская изодисомия, т. Е. Когда обе хромосомы 15 получены от матери. Развитие синдрома Ангельмана, наоборот, связано с микроделецией того же участка хромосомы 15, но материнского происхождения, или отцовской изодисомией. Большинство (около 70%) случаев синдрома Прадера — Вилли обусловлено микроделецией, остальные — дисомией. При этом обращает на себя внимание отсутствие клинических различий между больными с микроделецией и изодисомией.
Патогенез
Патогенез синдрома Прадера — Вилли до настоящего времени остается малоисследованным. Высказываются предположения, что ожирение у больных обусловлено значительным (более чем в 10 раз) усилением синтеза жира из ацетата и крайне низкими процессами липолиза.
Гипогонадизм по гипогонадотропному типу может быть связан с дисфункцией гипоталамуса, преимущественно, в области вентромедиального и вентролатерального ядер. Правильность данной точки зрения подтверждается эффективностью лечения больных фармацевтическими препаратами (кломифен), приводившими к увеличению в плазме содержания лютеинизирующего гормона, тестостерона, нормализации показателей почечной экскреции гонадотропинов, сперматогенеза и появлению вторичных половых признаков.
Одним из объяснений гипопигментации кожи, волос и радужки служит снижение активности тирозиназы в волосяных фолликулах и меланоцитах, а также уменьшение пигмента в сетчатке.
Обращается внимание на повышенный риск развития лейкемии у больных с синдромом Прадера — Вилли. Исследования выявили снижение репарации ДНК (до 65% по сравнению с 97% у здорового ребенка) в лимфоцитах больных с данной патологией. Не исключено, что низкая репарационная способность ДНК может играть роковую роль в развитии злокачественных новообразований у лиц с синдромом Прадера — Вилли.
Симптомы
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание.
В течение заболевания можно выделить две фазы: первая — свойственна детям 12-18 мес жизни. Она характеризуется выраженной мышечной гипотонией, снижением рефлексов — Моро, сосательного и глотательного, что затрудняет кормление ребенка. Вторая — наступает позже, через несколько недель или месяцев. Появляются полифагия, постоянное чувство голода, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей.
Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает. Стопы и кисти больных диспропорционально маленькие — акромикрия. У детей отмечается гипогонадизм (у мальчиков — гипоплазия полового члена, мошонки, крипторхизм, а у девочек — недоразвитие половых губ и в 50% случаев — матки).
Рост больных нередко снижен. У 75% детей наблюдается гипопигментация кожи, волос и радужки. Часто диагностируется микроцефалия. Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед. ). Речь затруднена, словарный запас уменьшен. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, страбизм.
Встречаются и другие аномалии: микродонтия, гипоплазия хрящей ушных раковин, сколиоз, эктропион (выворот века), глаукома.
Нередко развитие сахарного диабета, который с возрастом имеет тенденцию к улучшению.
При морфологическом исследовании мозга и ЯМР-томографии могут наблюдаться (примерно в 12% случаев) кисты червя мозжечка, аномалии коры головного мозга.
Продолжительность жизни больных может достигать 60 лет и более.
Лечение
Терапия синдрома Прадера — Вилли окончательно не разработана. По данным литературы, комплекс лечебных мероприятий включает лишь диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины).
Источник
Синдром Рассела-Сильвера является довольно редким врожденным заболеванием (1-30 случаев на 100 000 чел.). Патология диагностируется в раннем детском возрасте и характеризуется задержкой роста (в т.ч. внутриутробной), нарушением формирования костной системы, а также полового созревания. Рассмотрим причины, симптомы и особенности лечения данного недуга.
Синдром Рассела-Сильвера: особенности проявления
Заболевание было впервые описано педиатрами Сильвером и Расселом в середине ХХ века. Они в своих исследования выявили взаимосвязь между повышением гонадотропного гормона в организме человека и симптомом низкорослости.
Позднее было доказано влияние этого гормона на половое развитие.
Синдром Рассела-Сильвера диагностируется у лиц как женского, так и мужского пола. В большинстве случаев это генетическое расстройство встречается спорадически, хотя крайне редко встречаются родословные с данным недугом.
Причины
Точных данных о причинах развития синдрома Рассела-Сильвера на сегодняшний день нет.
Многочисленные исследования дают возможность сделать вывод, что здесь имеет место генетический провокатор процесса, который передается от матери.
Причиной преждевременного полового развития, которое происходит у пациентов с данным диагнозом, является переизбыток гонадотропных гормонов (ФСГ, ЛГ, ХГ, пролактин).
Симптомы
Синдром Рассела-Сильвера (мкб10) имеет характерные признаки проявления в младенческом возрасте:
- Небольшой вес при рождении.
- Увеличенная черепная коробка, выраженный лоб и узкий подбородок (создается впечатление «псевдогидроцефалии»).
- Недоразвитость половых органов.
- Узкий подбородок, маленький рот, уголки губ опущены.
- Позднее закрытие большого родничка.
- Недостаточный набор массы тела и роста.
Врожденный порок развития в первый год жизни ребенка часто дает о себе знать частой рвотой, запорами, гастроэзофагеальной болезнью.
Иногда первые симптомы болезни выражены нечетко, а позднее присоединяются такие нарушения:
- Ассиметрия строения тела, что ведет к нарушению походки.
- Сколиоз.
- Искривление V пальца.
- Низкий рост.
- Заболевания ЖКТ.
- Позднее прорезывание зубов.
- Кариес.
- Патологии почек.
- Наличие на кожных покровах округлых пятен кофейного оттенка разных размеров.
- Раннее половое созревание.
У детей с синдромом Рассела-Сильвера, как правило, рано появляются вторичные половые признаки (оволосение на лице у мальчиков, в паховой области и подмышками у обоих полов, менструации у девочек и т.п.). Интеллект таких больных, как правило, сохранен.
Диагностика
Современная медицина позволяет выявить данную патологию еще во время внутриутробного периода развития ребенка.
С 22-й недели беременности возможно проведение генетического исследования. Если анализ показывает возможность этого порока, имеется необходимость дифференцировать его с другими нарушениями:
- Синдром Фаркони.
- Синдром Блума.
- Синдром Неймеген.
Данные патологии имеют симптомы, схожие с синдромом Рассела-Сильвера, и являются не менее серьезными заболеваниями.
Будущих родителей предупреждают о возможных рисках, отклонениях в развитии ребенка, физических нарушениях, психологической травме как ребенка, так и его родственников, а также об отсутствии оптимального варианта коррекции состояния такого пациента.
Мать и отец должны принять решение о целесообразности дальнейшего вынашивания ребенка с диагнозом синдром Рассела-Сильвера.
Лечение
Данная болезнь до сих пор изучена недостаточно, поэтому едино правильной методики ее лечения не существует.
Исходя из этого, основной задачей в терапии синдрома является максимальное снижение влияния болезни на качество жизни человека, а также предупреждение возможных осложнений.
Раннее выявление патологии позволяет раньше начать корректировку ее развития.
Как правило, лечение синдрома Рассела-Сильвера включает прием таких гормональных средств:
- Хуматроп.
- Растан.
- Сайзен.
- Генотропин.
Целесообразность, дозировку и график приема данных препаратов определяет лечащий врач в индивидуальном порядке, отслеживая их влияние на состояние пациента.
Статистика показывает, что применение заместительной гормональной терапии приносит такие результаты:
- За первый год лечения пациент прибавляет в росте 8-13 см.
- За второй год больной вырастает еще на 5-6 см.
Кроме положительной динамики в росте человека, отмечается уменьшение ассиметрии строения тела, а также сколиоза.
Многие врожденные патологии развития, как и синдром Рассела-Сильвера, не имеют доказанной профилактической методики. Однако, уменьшить вероятность появления у ребенка заболеваний любого характера можно, если будущие родители будут соблюдать такие правила:
- До зачатия пройти медицинское обследование.
- Не принимать алкоголь и бросить курить за полгода до планируемой беременности.
- Не принимать лекарственные препараты без консультации с врачом.
- Изучить наследственную возможность генетических нарушений у ребенка.
- При наступлении беременности выполнять все рекомендации врача и беречь психологический комфорт будущей мамы.
Также во время вынашивания ребенка нужно стараться не допускать инфекционных заболеваний, поскольку многие из них, особенно на раннем сроке, могут вызывать значительные отклонения в развитии плода.
Синдром Рассела-Сильвера, к сожалению, является неизлечимой болезнью. Поэтому важно соблюдать все предписания врача и регулярно проходить все необходимые обследования. Это позволит проконтролировать и скорректировать состояние пациента на ранних этапах развития организма, а также полового созревания.
Поделиться ссылкой:
Реклама партнеров и статьи по теме
Источник
Современной медицине известно достаточно много заболеваний. Некоторые из них изучены довольно хорошо, над причинами и методами лечения других безуспешно работают группы ученых. Часть заболеваний приобретенные, другие же являются врожденными. Одним из таких врожденных заболеваний и является синдром Рассела-Сильвера.
Особенности заболевания
Существуют и другие популярные названия: карликовость Сильвера-Рассела, ССР.
Рассел А. и Х. К. Сильвер — врачи-педиатры, которые занимались изучением пренатальной задержки в развитии.
Синдром Рассела-Сильвера является заболеванием врожденным. Главной его особенностью является задержка физического развития еще во время беременности, в частности, нарушено формирование скелета ребенка. В дальнейшем может наблюдаться позднее закрытие родничка.
Причины наследования до сих пор неизвестны, в большинстве случаев нет определенной системы.
Данное заболевание встречается у одного человека на 30 000. Половая принадлежность на развитие болезни не влияет.
Причины возникновения синдрома Рассела-Сильвера
Главной причиной заболевания являются исключительно нарушения на уровне генетики. Причем форма наследования не носит периодический или системный характер.
Чаще всего страдают хромосомы 7 (10% случаев), 11, 15, 17, 18. Именно эти хромосомы и отвечают за рост человека. В большинстве случаев это происходит из-за того, что ребенок наследует две копии хромосомы от матери. Данный эффект носит название однородительской материнской дисомии.
Внешние симптомы заболевания
При рождении ребенок с синдромом Рассела-Сильвера имеет довольно маленькую массу, обычно не более 2500 г, хотя беременность и считается доношенной. Длина при этом около 45 см. С возрастом эта проблема не решается и отставание в росте наблюдается и у взрослых людей (у женщин рост не более 150 см, у мужчин чуть более 150 см). Однако масса полностью соответствует возрасту, в некоторых случаях даже превышает норму.

Претерпевает изменения и мочеполовая система, например, наблюдается крипторхизм (нарушение, при котором яички расположены не на своих местах), гипоспадия (мочеиспускательный канал открывается в нетипичном для этого месте), гипоплазия полового члена и мошонки (недоразвитость).
Внешне синдром Рассела-Сильвера также наблюдается. Выражается он в ассиметрии тела. Затрагивает это и лицо, и туловище, и длину ног и рук.
Влияет синдром Рассела-Сильвера (лечение заболевания можно узнать из статьи) и на лицо. Часть черепной коробки, в которой располагается головной мозг, увеличена по сравнению с лицевой ее частью, причем увеличение явно непропорционально. Форма лица напоминает треугольник, при котором лоб выпуклый, а размеры нижней челюсти и рта значительно уменьшены. Это называется псевдогидроцефалия. Губы узкие, а уголки слегка опущены (эффект «рот карпа»). Небо высокое, в некоторых случаях может быть с расщелиной. Уши в большинстве случае оттопырены.
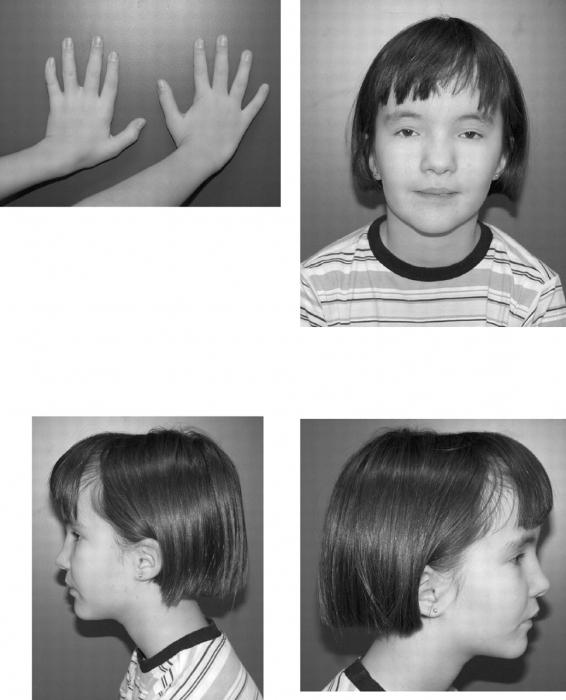
Среди сопутствующих внешних симптомов можно выделить:
- нарушение формирования подкожного жира;
- узкая грудная клетка;
- лордоз в области поясницы (выпуклость позвоночного столба вперед);
- искривления мизинца.
Сопутствующие заболевания внутренних органов
Помимо внешних расстройств, часто наблюдаются и внутренние проблемы организма. Синдром Рассела-Сильвера (симптомы, связанные с нарушениями внешнего вида, указаны ранее) влияет на работу почек в связи с их неправильной сформированностью (подковообразная форма, расширение почечной лоханки, ацидоз канальцев).
Для больных независимо от их половой принадлежности характерно раннее половое созревание. В 30% случаев начинается в возрасте около 6 лет. Это напрямую связано с тем, что происходит гонадотропная стимуляция яичников (количество половых гормонов значительно увеличено).
А вот интеллект полностью сохранен.
Синдром Рассела-Сильвера: диагностика
Диагностируется данное заболевание уже в раннем детстве. Такое решение принимает педиатр, наблюдающий больного ребенка. Однако помимо обычного наблюдения проводятся и различные лабораторные анализы и тесты:
- Определение уровня сахара в крови. Очень часто дети, которым ставится диагноз «синдром Рассела-Сильвера», имеют пониженный уровень глюкозы в крови.
- Тестирование на хромосомные аномалии. В большинстве случаев эти проблемы обнаруживаются.
- Определение количества гормона роста. При данном заболевании наблюдается его дефицит.
- Обследование сформированности скелета. Это требуется для того, чтобы исключить полностью дополнительные условия, которые в некоторых случаях могут давать ложный положительный результат.

Особенности лечения
Главное правило лечения: своевременная диагностика. Если не сделать этого вовремя, врач может пойти по ложному пути и заниматься лечением гидроцефалии, однако у таких детей этого заболевания нет.
В большинстве случаев таким больным назначается прием гормона роста по определенной схеме, которую разрабатывает лечащий врач.
Помимо этого часто используются и дополнительные методы:
- физиотерапия, которая направлена на улучшение состояния мышц;
- специальное образование.
В процессе лечения принимают одновременно несколько специалистов:
- врач-генетик, который способен выявить данное заболевание в самом его начале;
- диетолог или гастроэнтеролог, главное задание которого разработать особую диету, что направлена на повышение роста;
- эндокринолог, который и назначает гормон роста;
- психолог.
Определить эффективность проведенного лечения можно по увеличению скорости роста. При правильно разработанной схеме уже в первый год лечения можно достичь результата в 8 см.
Источник