Синдром секкеля по мкб 10

Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Секкеля — лечение в Италии
Синдром Секкеля (синонимы: карликовость с «птицеголовостью», примордиальная карликовость с микроцефалией) — редкое наследственное заболевание, характеризующееся пре- и постнатальным отставанием в росте, микроцефалией с задержкой психического развития и характерным изменением лица, напоминающим «птицеголовость».
Заболевание, по всей вероятности, было известно давно, так как термины «птицеголовая карликовость» и «наноцефалия» принадлежат Н.Зеске в 1960 г. опубликовал статью, в которой привел результаты обследования двух собственных пациентов и литературные данные о 24 больных, имевших сходные клинические проявления
Синдром Секкеля — редкий синдром, характеризующийся тяжелой низкорослостью, низким весом при рождении, очень маленькой головой (микроцефалия), большим лбом, большими глазами, низкими ушами, большим носом и небольшим подбородком. Дефекты костей рук и ног, вывихи локтевых суставов и бедер, неспособность выпрямлять колени и крипторхизм, также часто встречаются у детей с этим синдромом.
Хромосомная нестабильность и недопроизводство всех типов клеток крови (панцитопения) отмечаются у некоторых пациентов. Это заболевание является генетическим. Оно наследуется по аутосомно-рецессивному типу. Синдром Секкеля является неоднородным генетическим заболеванием, которое может быть связано с мутациями в генах, расположенных на хромосомах 3 и 18.
Следует отличать синдрос Секкеля от другиз заболеваний с низкорослостью. Для этого проводится дифференциальная диагностика при характерных дополнительных симптомах.
Низкая масса тела при рождении в случае доношенной беременности, внутриутробная задержка роста. Этот частый симптом свидетельствует о наличии генетически обусловленного снижения потенции роста тканей, недостаточности плаценты или же нарушений роста во внутриутробном периоде токсического или воспалительного генеза. Если отсутствует дополнительная симптоматика и вместе с тем имеющуюся клиническую картину нельзя отнести к какому-либо определенному заболеванию, говорят о примордиальной низкорослости. При отсутствии других симптомов последняя описывается как семейное заболевание.
В спорадических случаях следует думать о необратимом внутриутробном повреждении редупликационного потенциала клеток неизвестным патогенным фактором. При чисто примордиальной низкорослости дифференцировка скелета соответствует хронологическому возрасту.
- а) Примордиальная низкорослость: синдром Рассела — Сильвера.
- б) Синдром Секкеля: гетерогенная группа примордиальных карликов с микроцефалией, большим носом, скошенным подбородком и интеллектуальным недоразвитием.
- в) Прогерия (синдром Гатчинсона-Джилфорда): замедление роста и развитие начиная со 2-3-го года жизни, преждевременное старение, атрофия кожи, выпадение волос.
- г) Синдром Блума: телеангиэктатическая эритема щек, долихоцефалия.
- д) Синдром Корнелии де Ланге; умственное недоразвитие, густые, сросшиеся над переносицей брови, микроцефалия, усиленное оволосение тела.
- е) Лепрехаунизм (синдром Донохуа): отсутствие или недоразвитие подкожной жировой клетчатки, гипертрофия клитора или полового члена, усиленное оволосение.
- ж) Синдром Фанкони: панцитопения, гипоплазия I пальца руки или нарушение его закладки, пигментные аномалии.
- з) Семейная, или полигенная, низкорослость, при которой в большинстве случаев наблюдается только легкая задержка роста во внутриутробном периоде.
- и) Трисомия 18.
Очень короткие конечности (увеличенное отношение верхнего сегмента к нижнему). К этой группе прежде всего относятся заболевания скелета, обусловленные поражением эпифизов и метафизов. При дифференциальной диагностике обязательно следует подумать об атиреозе или тяжелом гипотиреозе (особенно учитывая возможности их лечения). Фосфатный диабет также может сопровождаться легкой диспропорциональной низкорослостью с преобладанием верхнего сегмента. Пропорции могут существенно меняться в процессе роста. Здесь представлены сведения о пациентах старшего детского и юношеского возраста в соответствии с обобщением Спрангера.
- а) Ахондроплазия (хондродистрофия): относительно большая голова с провалившимся основанием носа, особенное укорочение предплечий и голеней, кифоз, типичные рентгенологические находки.
- б) Гипохондроплазия: слабо выраженная диспропорциональная низкорослость, переразгибание суставов, люмбальный лордоз, типичные рентгенологические данные.
- в) Псевдоахондроплазия: пропорции, как и при ахондроплазии, но с нормальным формированием мозгового и лицевого черепа, относительно позднее по сравнению с ахондроплазией проявление заболевания, отсутствие типичных рентгенологических симптомов со стороны таза.
- г) Синдром Эллиса-Ван-Кревельда: порок сердца, дистрофия ногтей и пальцев стоп, эписпадия.
- д) Синдром Маркезани: изменения хрусталиков, как при синдроме Марфана (дрожание хрусталиков, может быть шаровидный хрусталик), брахидактилия, возможны легкие контрактуры.
- е) Метафизарная хондродисплазия, тип Шмидта: укорочение трубчатых костей (больше проксимальных), грибовидно вздутые эпифизы с размытыми очертаниями.
- ж) Метафизарная хондродисплазия, тип Мак-Кусика: редкие тонкие волосы, брови и ресницы, переразгибание суставов, типичная рентгенологическая картина.
- з) Метафизарная хондродисплазия, тип Енсена: утолщенные суставы с костным ограничением подвижности, формирование косолапости стоп, типичная рентгенологическая картина.
- и) Дисхондростеоз (синдром Лери — Вейля): укорочение предплечья и лучевой кости по отношению к локтевой, дорсальный, поддающийся редрессации подвывих дистального конца локтевой кости (деформация Маделунга).
- к) Пикнодизостоз: короткие концевые фаланги, большой мозговой череп, большие роднички (которые могут быть открытыми до зрелого возраста), спонтанные переломы.
- л) Карликовость с микромелией: гипоплазированные локтевая и малоберцовая кости, а также нижняя челюсть.
- м) Карликовость с микромелией: характерная «треугольная» деформация большеберцовой кости.
- н) Псевдогипопаратиреоз: ожирение, обращающее на себя внимание круглое лицо, укороченные пальцы рук, изменения метаболизма.
- о) Псевдопсевдогипопаратиреоз.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку — так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 –
срочное лечение в Италии
ЗАПРОС в КЛИНИКУ
Источник
Содержание
- Описание
- Дополнительные факты
- Симптомы
- Диагностика
- Причины
- Профилактика
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Болезнь Келера.
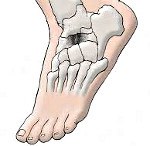
Болезнь Келера
Описание
Болезнь Келера. Хроническое дистрофическое заболевание костей стопы, приводящее к их асептическому некрозу. Заболевание может протекать с поражением ладьевидной кости (болезнь Келера I) или плюсневых костей (болезнь Келера II). Болезнь Келера проявляется отечностью и болями в стопе в области пораженной кости, усилением болевого синдрома при ходьбе и его прогрессированием с течением времени, изменением походки и хромотой при одностороннем поражении. Характерным признаком является отсутствие воспалительных изменений пораженной области. Диагностика заболевания основывается на данных рентгенологического исследования. Лечение состоит в снятии нагрузки с пораженной стопы путем ее иммобилизации и последующей восстановительной терапии (лечебная гимнастика, физиотерапия, массаж).
Дополнительные факты
Заболевание, описанное Келером в 1908 году, получило название болезнь Келера I. Оно представляет собой асептический некроз ладьевидной кости, в то время как в травматологии известна также болезнь Келера II — асептический некроз головок костей плюсны. Дегенеративно-дистрофические процессы в костной ткани, лежащие в основе болезни Келера, послужили основанием для ее причисления к группе остеохондропатий, куда также входят болезнь Кальве, болезнь Тиманна и болезнь Шляттера. Болезнь Келера встречается в основном в детском возрасте. Наиболее часто болезнь Келера I наблюдается у мальчиков от 3 до 7 лет, а болезнь Келера II — у девочек 10-15 лет.
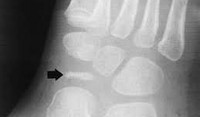
Болезнь Келера
Симптомы
Симптомы болезни Келера I.
Болезнь Келера I характеризуется появлением на тыльной стороне стопы ближе к ее внутреннему краю припухлости, обусловленной отечностью тканей этой области. Отсутствие покраснения кожи и местного повышения температуры в области отека свидетельствует в пользу невоспалительного характера происходящих изменений. Отмечается болезненность пораженной области при прощупывании и при нагрузке на стопу, утомляемость ребенка при ходьбе. Чтобы избежать боли при ходьбе, дети, имеющие болезнь Келера, ставят ногу с упором на наружный край стопы. Может наблюдаться хромота. Со временем боль усиливается и приобретает постоянный характер, не исчезая даже при полном покое. Болезнь Келера I длится в среднем около года и может привести к стойкой деформации ладьевидной кости.
Симптомы болезни Келера II.
Болезнь Келера II проявляется припухлостью и болезненностью в области пораженной плюсневой кости. Чаще всего встречается поражение II и III плюсневых костей. Возможен двусторонний характер патологических изменений. При этом симптомы воспаления не наблюдаются. Болезнь Келера II начинается с появления неинтенсивного болевого синдрома, поначалу проявляющегося лишь при нагрузке на передние отделы стопы. Характерно усиление боли при прощупывании пораженной области и во время ходьбы, особенно по неровному грунту или в обуви со слишком тонкой и мягкой подошвой. Со временем пациенты жалуются на то, что боль в стопе становиться постоянной, более интенсивной и сохраняется даже в покое. Отмечается укорочение пальца, который примыкает к головке подвергшейся некрозу плюсневой кости. Объем движений в суставе, сформированном пораженной плюсневой костью, ограничивается. Болезнь Келера II протекает в среднем в течение 2-3 лет.
Боль в голеностопе.
Диагностика
Диагностика болезни Келера I.
Болезнь Келера I диагностируется рентгенологически. На рентгенограммах стопы в начале заболевания отмечается остеопороз ладьевидной кости, вызванный асептическим разрушением ее губчатого вещества. Затем выявляется уплотнение точек окостенения, сплющивание и уплотнение ладьевидной кости. Еще позже наблюдается дефрагментация ладьевидной кости, т. Е. Ее распад на отдельные костные фрагменты в результате прогрессирования некротического процесса.
Диагностика болезни Келера II.
Диагностика заболевания основана на рентгенологическом исследовании стопы, в ходе которого выявляются патологические изменения в головке пораженной плюсневой кости. В зависимости от срока заболевания может наблюдаться остеопороз, уплотнение и деформация головки плюсневой кости, ее патологический перелом и дефрагментация.
Причины
Как и этиология других остеохондропатий факторы, вызывающие болезнь Келера, пока окончательно не изучены. Большинство исследователей склонны считать основной причиной некротических изменений костной ткани нарушение ее питания за счет расстройства местного кровоснабжения. Нарушения васкуляризации кости в свою очередь могут быть обусловлены врожденными особенностями кровообращения этой области, наличием поперечного или продольного плоскостопия, ношением неудобной или слишком тесной обуви, повторными травмами: ушибами, подвывихами или вывихами стопы, переломами костей стопы Роль благоприятствующих факторов в развитии болезни Келера могут играть различные обменные нарушения и эндокринопатии (гипотиреоз, сахарный диабет, ожирение).
Профилактика
Предупредить болезнь Келера у ребенка поможет правильный подбор обуви, которая должна соответствовать размеру ноги, быть удобной и не слишком жесткой. Следует избегать травм стопы, а при их получении незамедлительно обращаться к травматологу и следовать всем его рекомендациям. Поскольку болезнь Келера связана с плоскостопием, то его своевременное лечение также имеет профилактическое значение.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: M94.0
МКБ-10 / M00-M99 КЛАСС XIII Болезни костно-мышечной системы и соединительной ткани / M80-M94 Остеопатии и хондропатии / M91-M94 Хондропатии / M94 Другие поражения хрящей
Определение и общие сведения[править]
Синдром Титце
Синдром Титце — относительно редкое состояние, характеризующееся наличием неспецифического доброкачественного обратимого болезненного отека в области II (в 60% случаев) или III реберных хрящей. В 80% случаев поражение одностороннее и ограничено одним реберным хрящом.
Обычно заболевание развивается в молодом или детском возрасте.
Синдром Титце впервые описан А. Tietze в 1921 г.
Этиология и патогенез[править]
Причины его не известны, но у большинства пациентов в анамнезе выявляют предшествующие эпизоды респираторных инфекций, сильного кашля, тяжелой физической нагрузки, а также недостаточное питание.
Клинические проявления[править]
Боль обычно хорошо локализована, но может иррадиировать по всей передней поверхности грудной стенки, а также в надплечье и шею. Покраснение, повышение температуры и другие изменения кожи над областью поражения отсутствуют. Боль обычно регрессирует спонтанно через 2-3 нед, но часто продолжает беспокоить на протяжении нескольких месяцев, а резидуальный отек может сохраняться до нескольких лет.
Синдром хрящевых реберных соединений (Титце): Диагностика[править]
Дифференциальный диагноз[править]
Синдром хрящевых реберных соединений (Титце): Лечение[править]
Патогенетического лечения не существует, заболевание имеет доброкачественное самоограничивающееся течение, о чем необходимо проинформировать пациента. При выраженной боли возможно назначение НПВС внутрь или местно в виде мазей, физиопроцедуры
с фонофорезом глюкокортикоидов на пораженную область, ее согреванием или охлаждением.
Профилактика[править]
Прочее[править]
Реберно-грудинный синдром
Синонимы: синдром передней грудной стенки, костохондрит, реберногрудинная хондродиния
Определение и общие сведения
Синдром Титце часто путают со значительно более распространенным реберно-грудинным синдромом — одной из наиболее частых причин болей в грудной клетке. Заболевание наиболее часто диагностируют у женщин после 40 лет, его патогенез остается неизвестным.
Клинические проявления
В отличие от синдрома Титце при реберно-грудинном синдроме пальпаторно в 90% случаев можно выявить множественные зоны болезненности: в парастернальной области, ниже молочной железы, в проекции грудных мышц и грудины. Локальный отек отсутствует. Наиболее часто поражаются хрящи II и V ребра. При поражении верхних реберных хрящей боль обычно иррадиирует в область сердца. Она обычно усиливается при движениях грудной клетки.
Дифференциальный диагноз
Реберно-грудинный синдром дифференцируют с ИБС, при этом кроме особенностей болей, которые обычно носят не типичный для ИБС характер, также применяют блокады межреберных нервов с введением местных анестетиков по задней подмышечной линии, приносящие пациентам выраженное облегчение.
Лечение
Лечение симптоматическое. Показано назначение НПВС, парацетамола, при недостаточной их эффективности — в сочетании со слабыми опиоидными анальгетиками. Можно использовать физиотерапевтическое воздействие — согревание болезненной области, чрескожную электронейростимуляцию. Применяют блокады межреберных нервов с местными анестетиками по задней подмышечной линии. Эта манипуляция имеет и психотерапевтическое значение, поскольку регресс болевого синдрома после ее проведения позволяет убедить пациента в доброкачественности заболевания и его «некардиогенной» причине.
Источники (ссылки)[править]
Боль в спине [Электронный ресурс] / Подчуфарова Е.В. — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970424742.html
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Рубрика МКБ-10: Q79.6
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q65-Q79 Врожденные аномалии пороки развития и деформации костно-мышечной системы / Q79 Врожденные аномалии (пороки развития) костно-мышечной системы, не классифицированные в других рубриках
Определение и общие сведения[править]
Синдром Элерса-Данло (Ehlers-Danlos) — генетически гетерогенное заболевание, обусловленное разнообразными мутациями в генах коллагена, либо в генах, отвечающих за синтез ферментов, принимающих участие в созревании волокон коллагена. Характеризуется гиперэластичностью кожи, подкожными сферулами, переразгибанием суставов, лёгкой ранимостью тканей и геморрагическим синдромом.
Эпидемиология
Истинная распространённость неизвестна вследствие сложности верификации и большого числа лёгких форм, частота диагностированных случаев — 1 на 5000 новорождённых, тяжёлые формы встречают редко (1:100 000).
Классификация
В генетической классификации описано 11 типов и ряд подтипов с разным наследованием, клиническими особенностями и биохимическими дефектами.
Этиология и патогенез[править]
Синдром Элерса-Данло — группа заболеваний соединительной ткани, различающихся по типу наследования, клиническим особенностям и биохимическому дефекту. В большинстве случаев наследуется по аутосомно-доминантному типу, сопровождается уменьшением количества или изменением структуры коллагена.
Клинические проявления[править]
Синдром Элерса-Данло: Диагностика[править]
Объём обследования определяется наличием ведущих клинических признаков заболевания. Существенное значение имеет генеалогическое исследование и молекулярно-генетические методы диагностики.
Диагностику СЭД проводят согласно Вилльфраншской классификации (Beighton P. et аl., 1998).
Для диагностики СЭД необходимо выполнение следующих требований.
• Для клинической диагностики необходимо наличие хотя бы одного большого критерия. При соответствующих возможностях наличие одного или более больших критериев гарантирует подтверждение СЭД на лабораторном уровне.
• Малый критерий — признак, обладающий меньшим уровнем диагностической специфичности. Наличие одного или более малых критериев вносит свой вклад в диагностику того или иного типа СЭД.
• При отсутствии больших критериев для установления диагноза малых недостаточно. Наличие малых критериев даёт основание предполагать наличие состояния, подобного СЭД, характер которого будет разъясняться по мере того, как станет известной его молекулярная основа. Поскольку встречаемость малых критериев существенно выше, чем больших, в полном согласии с Вилльфраншским пересмотром наличие только малых критериев даёт основание для диагностики элерсоподобного фенотипа.
Критерии диагностики СМ и СЭД включают гипермобильность суставов.
В случае невыполнения соответствующих критериев гипермобильность необходимо рассматривать как самостоятельное состояние.
Дифференциальный диагноз[править]
Синдром Элерса-Данло: Лечение[править]
Принципы лечения
Богатая белком диета, содержащая костные бульоны, студни, заливные блюда. Курсы массажа, физиотерапии, лечебная физкультура. Посиндромная терапия, зависящая от выраженности органных изменений. Медикаментозное лечение с использованием аминокислотных (карнитин, витаминных (витамины D, С, Е, В1, В2, В6), минеральных комплексов (кальция карбонат/холекальциферол, магнерот), хондроитина сульфата перорально и местно, глюкозамин, оссеин-гидроаппатитных комплексов (остеокеа, остеогенон), трофических препаратов (АТФ, инозин, лецитин, коэнзим Q10). Указанные препараты принимают сочетанными курсами 2-3 раза в год продолжительностью 1-1,5 мес
Профилактика[править]
Прочее[править]
Прогноз
Прогноз благоприятный, серьёзнее при I (вследствие артропатий) и IV (вследствие кровотечений и разрывов сосудов) типах СЭД. Детей следует ориентировать на выбор профессии, не связанной с физическими нагрузками, работой стоя.
Источники (ссылки)[править]
Педиатрия [Электронный ресурс] / Под ред. А.А. Баранова — М. : ГЭОТАР-Медиа, 2009. — https://www.rosmedlib.ru/book/ISBN9785970410851.html
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник