Синдром с монголоидный разрез глаз
Первую и вторую группу дизморфий описывают как стигмы (stigma, stigmatos), что в переводе с греческого означает метка, знак. Для определения уровня стигматизации учитывают общее количество стигм у ребенка, независимо от их локализации. Превышение критического уровня Стигматизации (5-6 стигм, особенно II группы) указывает на диспластическнй тип развития и может расцениваться как вероятность наличия аномалии развития внутренних органов. Ряд стигм для одних детей является дизморфией, а для других — отражением нормального генетического развития данной семьи. Например, монголоидный разрез глаз является стигмой, если семья ребенка не относится к представителям монголоидной расы.
Чаще всего стигмы описывают по частям тела. Череп. Изменение размеров и формы головы, нависающая затылочная кость, плоский затылок, прямая линия скошенного лба, нависание лба, высокая (низкая) линия роста волос на затылке, лбу.
При изменении размеров головы выделяют микроцефалию (окружность головы менее 3 % перцентиля для возраста и пола) и макроцефалию (окружность головы больше 97 % перцентиля для возраста и пола). Разнообразные варианты формы головы подробно описаны в разделе «исследование костной системы». Следует подчеркнуть, что как стигмы дизэмбриогенеза любые изменения черепа могут рассматриваться только при отсутствии патологии. Так, например, при гидроцефалии отмечается выступающий лоб и сглаженный затылок, что является признаками этого заболевания, а не стигмами.
Выступающий лоб, сглаженный затылок и макроцефалия при гидроцефалии
Наряду с этим у ребенка имеются большие размеры головы, относительно маленький лицевой череп, расширение подкожных вен на голове, расхождение швов черепа, выбухание родничка.
Лицо. Искривление носа, широкая переносица, резко выраженные надбровные дуги, синофриз, прогнатия, микрогнатия, прогения, микрогения, изменения подбородка (раздвоение, скошенность, клиновидность).
Синофриз — сращение бровей в надпереносье. При нарушении развития верхней челюсти говорят о прогнатии (чрезмерное развитие и выступание вперед) или микрогнатии (обратное состояние). При нарушениях развития нижней челюсти говорят о прогении (чрезмерное развитие и выступание вперед) или микрогении (обратное состояние). На рисунке представлен один из вариантов изменения подбородка — клиновидный подбородок:
Клиновидный подбородок
Глаза. Монголоидный или антимонголоидный разрез глаз, гипертелоризм, гипотелоризм, микрофтальмия, макрофтальмия, асимметрия глазных щелей, колобома радужки, гетерохромия радужки, неправильная форма зрачков, эпикант, дистихиаз и другие нарушения роста ресниц.
При монголоидном разрезе глаз отмечаются узкие глазные щели, наружные углы глаз приподняты кверху, при антимонголоидном разрезе наоборот, наружные углы глаз опущены вниз. Для оценки расстояния между внутренними краями глазниц рассчитывают индекс межорбитальной окружности:
Расстояние между орбитами на уровне внутреннего угла глаза
При величине индекса меньше 3,8 % говорят о гипотелоризме, больше 6,8 % — о гипертелоризме
Гипертелоризм
Микрофтальмия — уменьшение, а макрофтальмия — увеличение всех размеров глазного яблока при отсутствии пороков развития глаз. Эпикант — вертикальная, полулунная кожная складка, прикрывающая медиальный угол глаза (третье веко).
Третье веко, энтропион
При наличии дефекта части радужки говорят о колобоме, при неодинаковой окраске радужки одного или правого и левого глаза — о гетерохромии радужки. Дистихиаз — двойной рост ресниц. Энтропион — заворот края нижнего века внутрь. Эктропион — выворот края нижи его века кнаружи.
Уши. Высоко (низко) расположенные уши, асимметрия расположения ушных раковин, большие оттопыренные уши, малые деформированные уши, разновеликие уши, приращенные мочки, отсутствие мочек, добавочные козелки, аномалии развития завитка и противозавитка.
Для определения расположения ушей пользуются горизонтальной линией, соединяющей углы глаза. Если верхняя часть ушной раковины присоединяется к голове ниже этой линии, говорят о низко расположенных ушах.
Низко расположенные уши
Если нижняя часть козелка уха расположена выше этой линии, говорят о высоком расположении ушей.
Асимметрия ушных раковин определяется при осмотре ребенка сзади.
Асимметрия расположения ушных раковин, низкий рост волос на затылке
Диагностика деформации и размеров ушей в основном строится на субъективном представлении.
Большие деформированные оттопыренные уши
Рот. Макростомия, микростомия, готическое нёбо, короткое нёбо, короткая уздечка языка, макроглоссия, микроглоссия, складчатый язык, раздвоенный кончик языка, раздвоенность.
Макростомия — большой рот, микростомия — маленький рог. Готическое нёбо — высокое, узкое нёбо. При больших размерах языка говорят о макроглоссии, при малых — о микроглоссии.
Большой язык, не помещающийся в ротовой полости
Зубы. Редкие зубы, сверхкомплектные зубы, неправильная форма (пилкообразные или шиповидные) или расположение зубов.
Шея и туловище. К о р о т к а я или втянутая шея, низкий рост волос на затылке, птеригиум, асимметрия или добавочные соски молочных желез, недоразвитие мечевидного отростка грудины, расхождение прямых мышц живота, неправильное расположение пупка, грыжи.
Птеригиум — крыловидные складки кожи на боковых поверхностях туловища.
Кисти и стопы. Широкая или короткая ладонь, поперечная борозда ладони, арахнодактилия, девиация, брахидактилия, клинодактилия, синдактилия, полидактилия, эктродактилия, камптодактилия, сандалневидная щель, нахождение пальцев стоп друг на друга, плоскостопие.
Поперечная складка ладони довольно часто встречается при хромосомной аномалии (болезнь Дауна), хотя может отмечаться и как изолированная стигма дизэмбриогенеза.
Поперечная борозда ладони
Арахнодактилия — длинные, «паучьи» пальцы.
Арахнодактилия
Брахидактилия — укорочение пальцев за счет недоразвития фаланг. При латеральном или медиальном искривлении пальцев говорят о клинодактилии, при полном или частичном сращении соседних пальцев — о синдактилии. При нарушении количества пальцев говорят о полидактилии (дупликации) — лишние пальцы — или об эктродактилии — уменьшение числа пальцев. Камптодактилия — сгибательная контрактура пальцев. Сандалиевидная щель — углубленный и расширенный промежуток между первым и вторым пальцем стоп.
Кожные покровы. Обилие депигментир о ванных или пигментированных участков, большие родимые пятна (с оволосением или без него), избыточное локальное оволосение.
Следует подчеркнуть, что подходы к оценке уровня стигматизации должны быть индивидуально ориентированными. Говорить о стигмах, в их истинном понимании, можно только при отсутствии пороков развития, генетической патологии. При наличии же последних стигматизация ребенка лишь подтверждает наличие неблагоприятного воздействия в период закладки и формирования органов и систем. В качестве примера можно привести ребенка со значимым «уровнем стигматизации» — большие оттопыренные и низко расположенные уши с нарушением их хрящевой структуры, гипертелоризм, высокая линия роста волос в области лба, нарушение расположения зубов, измененная форма черепа — всего 8 стигм только в области головы, а с учетом других стигм уровень стигматизации ребенка очень высокий — 12 стигм. При этом ребенок не имеет генетической патологии, врожденных пороков развития и по своим параметрам соответствует календарному возрасту.
Множественные стигмы дизэмбриогенеза
Источник
Аномалии глазной щели — варианты
а) Направление глазной щели. Углом или наклоном глазной щели называется угол, образованный линией, соединяющей внутреннюю и наружную спайки, и горизонталью. В норме глазная щель скошена несколько кверху. Разрез глаз описывается как монголоидный, если наружная спайка находится выше, чем обычно, и как антимонголоидный, если наружная спайка расположена ниже обычного.
Монголоидный разрез глаз может быть связан с микроцефалией. При трисомии 21 монголоидный разрез глаз — наиболее частый глазной или лицевой признак. Антимонголоидный разрез глаз часто наблюдается при гипоплазии скуловых костей. Также это характерный симптом при мальформациях первой или второй жаберных дуг, например, при синдроме Тричера Коллинза, который характеризуется узким лицом, гипоплазией надглазничного края, скул, гипоплазией ушей.
Глазная щель может иметь «волнистую» форму при синдроме Коэна, включающего в себя характерные изменения лица, задержку развития и дегенерацию сетчатчки.
б) Длинная глазная щель. Глазная щель считается длинной, если расстояние между внутренней и наружной спайками век на 2 SD превышает возрастную норму.
Эуриблефарон — это общее увеличение глазной щели, обычно больше с латеральной стороны. Имеется локальное смещение наружной спайки кнаружи и вниз и смещение нижнего века книзу. При поверхностном осмотре это может напоминать врожденный эктропион (выворот всего нижнего века). Эуриблефарон может возникать как изолированная аномалия, может передаваться по аутосомно-доминантному механизму или быть связанным с трисомией 21 или с краниофациальным синостозом.
Эуриблефарон характерен для синдрома кабуки: задержка роста в постнатальном периоде, умственная отсталость и черты лица, напоминающие грим актеров традиционного японского театра.
в) Короткая глазная щель. Глазная щель считается короткой, если расстояние между наружной и внутренней спайками составляет менее 2 SD по сравнению с возрастной нормой.
Умеренное укорочение длины глазной щели может быть следствием увеличенной кривизны края века («миндалевидный» разрез глаз) и наблюдаться при трисомии 21.
Блефарофимозом называется уменьшение максимального вертикального расстояния между верхним и нижним веками в сочетании с короткой глазной щелью. Блефарофимоз может быть изолированным или быть частью различных синдромов. Его нельзя путать с птозом (при котором глазная щель не укорочена).
Алкогольный синдром плода включает в себя задержку роста, микроцефалию и когнитивные нарушения. Это одна из наиболее частых причин блефарофимоза.
Синдром блефарофимоза — птоза — epicanthus inversus (BPES) — аутосомно-доминантное заболеванияе с выраженным блефарофимозом, птозом, гипоплазией тарзальных пластинок и epicanthus inversus. Описаны два клинических типа BPES:
1. BPES тип I: передача по мужской линии, нерегулярные месячные и бесплодие у больных женщин.
2. BPES тип II: не связан с бесплодием, передача через родителей обоего пола. Ранние вехи развития могут появляться с задержкой из-за гипотонии и запрокинутой назад головы.
Синдром Охдо обычно возникает спорадически и включает в себя блефарофимоз, птоз, гипоплазию зубов, частичную глухоту и умственную отсталость. Птоз и/или блефарофимоз также наблюдаются при хромосомных болезнях. Блефарофимоз и птоз, например, являются характерными признаками делеции 3p хромосомы.
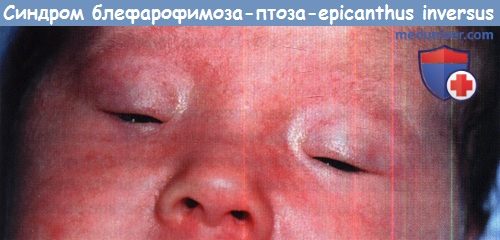
Синдром блефарофимоза-птоза-epicanthus inversus у двухмесячного ребенка.
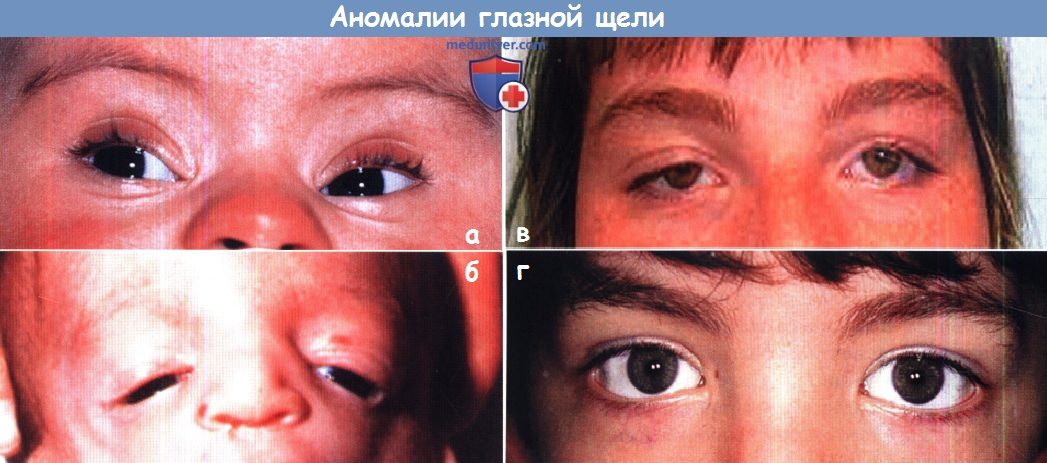
а — Монголоидный разрез глаз у ребенка с трисомией 21.
б — Антимонголоидный разрез глаз у ребенка с синдромом Тричера-Коллинза.
в — «Волнистая» глазная щель при синдроме Коэна (сопровождается дистрофией сетчатки).
г — Эуриблефарон при синдроме кабуки.
— Также рекомендуем «Аномалии положения век — варианты»
Оглавление темы «Аномалии век, бровей и ресниц у детей.»:
- Аномалии развития периорбитальной области век
- Аномалии внутренней спайки век и грубые аномалии век
- Аномалии глазной щели — варианты
- Аномалии положения век — варианты
- Варианты и классификация птоза — опущения века
- Аномалии бровей и ресниц — варианты
- Советы по лечению врожденных аномалий век
- Советы по лечению птоза — опущения века
- Лечение ретракции век новорожденных
- Лечение последствий паралича лицевого нерва у детей
Источник
Синдром Ди Джорджи получил свое название от имени открывшего его педиатра-эндокринолога. Иначе эта болезнь еще называется синдромом Ди Георга или велокардиофациальным синдромом. Заболевание редкое, в равной степени встречается у девочек и у мальчиков.
Болезнь Ди Джорджи характеризуется частичной либо полной гипоплазией вилочковой железы, которая находится в переднем средостении и отвечает за Т-клеточное звено иммунитета, и проявляется иммунодефицитным состоянием, мальформацией лицевой части черепа и врожденными пороками сердца. Кроме гипоплазии тимуса (недоразвития), данный синдром в себе часто сочетает отсутствие паращитовидных желез, что приводит к стойкому снижению уровня кальция в крови.
Т-лимфоциты – это клетки крови, вырабатывающиеся лейкоцитарным ростком красного костного мозга и в дальнейшем мигрирующие в вилочковую железу. Бывают нескольких типов: хелперы, супрессоры и киллеры. Они обеспечивают распознавание чужеродных белков (вирусы, грибки и бактерии), их устранение и предупреждают развитие аутоиммунной патологии (когда собственный организм уничтожает свои здоровые клетки и ткани).
Фото синдрома Ди Джорджи
В чем причина болезни Ди Джорджи?
Примерно у 85-90% больных, страдающих этой патологией была выявлена делеция 22 хромосомы – выпадение небольшого участка генетического материала.
К факторам риска, способных привести к такой генетической аномалии относят: прием алкогольных напитков и курение в течение беременности, перенесенные инфекции, особенно в первые три месяца развития плода и наличие у матери сахарного диабета.
Симптомы заболевания
Клинические проявления и симптомы велокардиофациального синдрома достаточно разнообразны и видны с самого рождения ребенка.
Наиболее ранним симптомом Ди Джорджи синдрома являются судороги, которые чаще всего возникают в первые несколько суток с момента появления ребенка на свет и даже могут послужить причиной смерти. Они обусловлены значительным снижением кальция в периферической крови из-за отсутствия паращитовидных желез.
Вследствие недоразвития вилочковой железы иммунная система таких детей не способна в полной мере дать адекватный ответ на внедрение в организм бактерий, вирусов и грибков, поэтому они в значительной мере подвержены инфекционным заболеваниям с тяжелым течением.
Обязательный симптом — врожденные пороки сердца. К примеру, тетрада Фалло, которая характеризуется:
- Сужением выводного сосуда правого желудочка – легочной артерии.
- Дефектом мышечного отдела перегородки между желудочками.
- Декстрапозицией аорты – отклонением ее ствола в правую сторону.
- Гипертрофией (увеличением стенки) правого желудочка.
Патология со стороны лицевого скелета:
- Расщелина неба и верхней губы.
- Гипертелоризм – патологически увеличенное расстояние между глазами.
- Высокое нёбо у ребенка или иначе — готическое небо.
- Деформация и низкое расположение ушных раковин.
- Антимонголоидный разрез глаз.
- Недостаточное развитие верхней челюсти в размере или микрогнатия.
Антимонголоидный разрез глаз – это опущение книзу внешнего угла глаза.
Антимонголоидный разрез глаз
Причиной готического неба является патологическое формирование эмбриональных закладок. Как таковых последствий у ребенка с высоким небом опасаться не стоит.
Фото готического неба у ребенка
Кроме того, такие дети имеют аномалии развития со стороны иных органов и систем.
Поражение со стороны нервной системы проявляется плохой координацией вследствие неполного развития мозжечка, расстройством чувствительных и моторных функций на фоне атрофии коры полушарий мозга.
У детей с синдромом Ди Джорджи часто выявляются укорочение пищевода, фистулы – патологические каналы в анальном отверстии, аномальное развитие сосудов сетчатки, гидронефротическая трансформация почек (скопление жидкости в почечной ткани) или их атрофия. Полидактилия (патологическое количество пальцев на руках или ногах), отсутствие ногтевой пластины и другие различные костные аномалии. Постоянный симптом – задержка интеллектуального развития.
Диагностика заболевания
В отличие от других делеционных заболеваний этот синдром можно выставить уже в родильном зале.
При осмотре новорождённого врач-неонатолог может отметить маленький размеры головы, антимонголоидный разрез глаз, страбизм или иначе — косоглазие, низко посаженные уши, широкую переносицу, расщелину мягкого нёба и верхней губы, а также иные дефекты, которые позволят заподозрить болезнь Ди Георга. Врач уже на раннем этапе при прослушивании сердечных тонов может выявить шумы, свойственные порокам.
Вышеуказанный синдром выставляется при сочетании таких симптомов: судороги с первых суток от рождения, врожденные пороки сердца, аномалии развития лицевого скелета, часто рецидивирующие грибковые и бактериальные инфекции, которые трудно поддаются терапии.
В течение нескольких недель повторно берутся анализы крови для определения количества Т-лимфоцитов, концентрации ионизированного кальция, количества иммуноглобулинов в сыворотке (те защитные белки, которые вырабатываются против бактерий, вирусов и т.д.). Исследования назначаются не единожды, чтобы убедиться в том, что снижение всех показателей имеет стойкий характер.
Обязательными методами в диагностике этой патологии являются УЗИ вилочковой и паращитовидных желез, эхокардиографическое исследование сердца (с целью выявления пороков).
Проводится и генетический анализ методом гибридизации ДНК для выявления участков выпадения в 22-й хромосоме.
Все указанные методы исследования позволяют не только точно определить саму болезнь, но и указать степень её тяжести, подобрать необходимое лечение.
Лечение синдрома Ди Джорджи
В данном случае излечить болезнь полностью невозможно, поэтому назначается комплексная поддерживающая терапия.
Для устранения инфекции, в зависимости от микроорганизмов, вызвавших ее, назначаются антибиотики, антимикотические (противогрибковые) и противовирусные препараты или их комбинации.
Так как у ребенка в огромном дефиците некоторые иммунные клетки – внутривенно либо внутримышечно вводится иммуноглобулин, получаемый из жидкой составной крови здоровых людей. Такая терапия называется заместительной.
С целью профилактики развития судорожного синдрома назначаются лекарственные препараты кальция.
Хирургическое лечение применяется для ликвидации аномалий развития лица и пороков сердца.
В отдельных случаях – при выраженном иммунодефиците, проводится трансплантация (т.е. пересадка) вилочковой железы. Забор органа производится от плода, внутриутробный возраст которого не превышает 14 недель. Пересадка может осуществляться на переднюю брюшную стенку в мышечную ткань либо клетки тимуса вводятся внутривенно в виде взвеси. Операция проводится в том случае, если пороки сердца были скорректированы.
Люди с таким заболеванием должны избегать переохлаждений, частых стрессовых ситуаций, контакта с инфекционными больными.
Как таковой профилактики для синдрома Ди Джорджи не существует. С целью предупреждения развития патологий у плода женщина, планирующая беременность, может сделать прививки против таких вирусных болезней, как корь и краснуха, так как они обладают сильным тератогенным действием на эмбрион. Беременная должна категорически отказаться от курения и употребления алкогольных напитков, избегать инфекционных заболеваний.
Будущая мать также может сделать специальный пренатальный тест, позволяющий выявить генетические отклонения у плода и при наличии таковых вместе с лечащим доктором обсудить исход беременности.
Возможные последствия и осложнения болезни
- Выраженная задержка в умственном развитии.
- Появление опухолевых заболеваний, в том числе и злокачественных.
- Возникновение различных аутоиммунных болезней.
- При тяжёлых инфекциях и врожденных пороках сердечно-сосудистой системы, возможен летальный исход.
Прогноз для жизни у таких детей относительно неблагоприятный, так как вследствие тяжелейшего иммунодефицита, они редко достигают возраста старше 6-7 лет.
Итак, синдром Ди Джорджи – это генетическое иммунодефицитное заболевание с тяжелыми аномалиями скелета лица и сердечно-сосудистой системы, которое чаще всего диагностируется в роддоме. Специфической профилактики и лечения не существует, возможна только поддерживающая терапия. Как правило, описанная патология имеет неблагоприятное течение.
Источник