Синдром рубинштейна тейби код мкб
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Рубинштейна-Тейби.
Синдром Рубинштейна-Тейби
Описание
Синдром Рубинштейна. Тейби — генетически гетерогенное (по последним данным) наследственное заболевание, характеризующееся поражением центральной нервной системы, деформациями костей скелета и рядом других пороков развития. Симптомами этого состояния являются прогрессирующая умственная отсталость, низкий рост, расширение фаланг пальцев, полидактилия на ногах, разнообразные нарушения со стороны внутренних органов. Диагностика синдрома Рубинштейна-Тейби производится на основании данных настоящего статуса пациента, молекулярно-генетических анализов и других исследований. Специфического лечения данной патологии не существует, применяют симптоматическую терапию в зависимости от типа пороков и нарушений.
Дополнительные факты
Синдром Рубинштейна-Тейби – генетическое заболевание, которое сопровождается нарушениями интеллектуального и физического развития, а также разнообразными пороками скелета и внутренних органов. Впервые данная патология была выявлена в 1963 году и описана американскими педиатрами Дж. Рубинштейном и Г. Тейби – сами исследователи назвали заболевание «синдромом широкого первого пальца кистей и стоп, специфического лица и умственной отсталости». Дальнейшие исследования в области генетики подтвердили, что синдром Рубинштейна-Тейби является аутосомно-доминантной патологией, но подавляющее большинство случаев возникает вследствие спонтанных мутаций различного типа. Встречаемость составляет порядка 1:100 000-125 000, мальчики и девочки поражаются примерно с одинаковой частотой. Особенностью синдрома Рубинштейна-Тейби является повышенный риск развития различных онкологических заболеваний – главным образом, некоторых типов лейкозов, опухолей мозговых оболочек и нервных тканей.
Синдром Рубинштейна-Тейби
Причины
Синдром Рубинштейна-Тейби обладает выраженной генетической гетерогенностью, однако различные типы этого заболевания не имеют каких-либо особенностей клинического течения. Наиболее часто причиной этой патологии выступают повреждения гена CREBBP, который расположен на 16-й хромосоме. Он кодирует так называемый CREB-связывающий белок, являющийся важным транскрипционным фактором, контролирующим экспрессию огромного количества генов. Появление в гене CREBBP различных изменений (точечных мутаций, транслокаций, микроделеций в 16-й хромосоме) приводит либо к образованию дефектного белка, неспособного выполнять свои функции, либо к полной блокировке его выделения. Это становится причиной развития синдрома Рубинштейна-Тейби.
В последние годы молекулярно-генетические исследования данной патологии подтвердили, что в ее развитии играют роль и мутации гена EP300, который располагается на 22-й хромосоме. Продуктом экспрессии данного гена является протеин p300, который, как и CREB-связывающий белок, относится к группе транскрипционных факторов. По данным исследований, идентифицировать характер генетического дефекта при синдроме Рубинштейна-Тейби удается в 55-60% случаев, примерно у половины пациентов обнаруживаются мутации гена CREBBP, еще у 40-45% выявляются делеции и другие хромосомные перестройки, затрагивающую 16-ю хромосому, и лишь у 3% больных наблюдаются мутации гена EP300. Все это указывает на возможность участия других генов в развитии этого заболевания.
Дефект белка-фактора транскрипции или его малое выделение (по причине гаплонедостаточности) приводит к многочисленным нарушениям, обусловленным недостаточной стимуляцией ряда других генов. Это можно заметить по характерной клинической картине синдрома Рубинштейна-Тейби. Оба транскрипционных фактора влияют на активность образования новых связей между нейронами, формирование долговременной памяти, стимуляцию иммунного ответа, развитие репродуктивной системы. Недостаточность этих белков ведет к целому каскаду патологических процессов в организме, что и составляет клинику синдрома Рубинштейна-Тейби. Кроме того, вышеуказанные транскрипционные факторы также влияют на активность антионкогенов, поэтому при их дефекте значительно возрастает риск развития злокачественных новообразований.
Симптомы
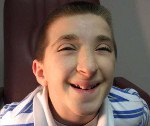
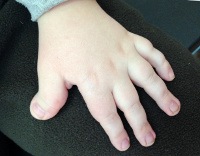
Симптоматика синдрома Рубинштейна-Тейби характеризуется значительным разнообразием у разных больных, что отражается на тяжести течения заболевания и его прогнозе. Обычно уже при рождении можно обнаружить некоторые признаки этой патологии – деформации лица и черепа (микро- или брахицефалия, расширение переносицы, эпикант, клювовидный нос), высокое арковидное нёбо, изменение формы и положения ушных раковин, расширенные фаланги пальцев, особенно первых. На ногах у больных синдромом Рубинштейна-Тейби нередко выявляется полидактилия. Иногда уже при рождении определяются признаки пороков развития внутренних органов – бледность или цианоз (при поражении легких или сердца), длительная желтуха новорожденных, крипторхизм.
При дальнейшем течении синдрома Рубинштейна-Тейби отмечается прогрессирующее отставание ребенка в физическом и умственном развитии от здоровых сверстников, затрудненное развитие речи и моторных навыков. Также наблюдается косоглазие, миопия. Выражение лица больных характеризуется гримасой, напоминающей улыбку, возникает гирсутизм, примерно у половины пациентов определяется наличие красного невуса в области лба, затылка или шеи. В старшем возрасте у больных синдромом Рубинштейна-Тейби выявляются искривления позвоночного столба различного характера и низкий рост (не более 150-160 сантиметров). Могут определяться деформации грудной клетки и (реже) другие скелетные аномалии, у мальчиков в большинстве случаев отмечается крипторхизм.
Постоянным признаком синдрома Рубинштейна-Тейби является умственная отсталость. Как правило, диагностируется ЗПР, олигофрения, значительная задержка речевого развития по сравнению со здоровыми сверстниками. Отмечаются нарушения концентрации внимания, больные легко отвлекаются на посторонние раздражители при выполнении какого-либо задания, возможны резкие перепады настроения. При этом лица с синдромом Рубинштейна-Тейби хорошо идут на контакт, легко социализируются. Среди других неврологических симптомов заболевания нередко обнаруживается плохая координация движений, изредка наблюдаются судорожные приступы.
Судороги.
Диагностика
Для выявления синдрома Рубинштейна-Тейби используют данные общего осмотра больного, рентгенологических исследований и молекулярно-генетических анализов, вспомогательную роль могут играть УЗИ и МРТ. При осмотре определяются характерные для этого заболевания аномалии развития лица (изменение формы и размеров черепа и носа, антимонголоидный разрез глаз), укорочение и расширение фаланг больших пальцев на руках и ногах, иногда – полидактилия. У взрослых больных синдромом Рубинштейна-Тейби также выявляют уменьшение роста, глубокую умственную отсталость, искривления позвоночника. На рентгенограммах можно увидеть изменения костей фаланг, позвоночника и грудной клетки, костный возраст несколько отстает от фактического.
Молекулярно-генетическая диагностика синдрома Рубинштейна-Тейби выполняется врачом-генетиком, который может использовать множество методов для определения этого заболевания. Точечные мутации в генах CREBBP и ЕР300 выявляются посредством прямого автоматического секвенирования кодирующей последовательности. Методика FISH используется в том случае, когда подозревается наличие делеций или транслокаций на 16-й хромосоме, также приводящих к развитию синдрома Рубинштейна-Тейби. Так как на основании фенотипических данных крайне сложно определить возможный тип генетического дефекта, в рамках диагностики этого заболевания нередко приходится применять сразу несколько техник современной молекулярной генетики.
Вспомогательные методы диагностики позволяют выявить сопутствующие нарушения внутренних органов при синдроме Рубинштейна-Тейби. К таким методам относят ультразвуковые исследования (УЗИ почек и мочевыделительных путей, ЭхоКГ), электрокардиографию, магнитно-резонансную томографию и другие методики. Нередко у больных синдромом Рубинштейна-Тейби диагностируют врожденные пороки сердца (открытый Боталлов проток, дефект межжелудочковой перегородки), аритмии, аномалии почек (удвоение, гипоплазия) и мочевыделительных путей (атрезии на различных участках). Также при этом заболевании нарушается формирование мозолистого тела головного мозга, что определяется при помощи магнитно-резонансной томографии. У части больных синдромом Рубинштейна-Тейби отмечаются пороки развития легких и пищеварительной системы.
Лечение
Специфического лечения данной патологии на сегодняшний момент не существует, все терапевтические мероприятия сводятся к облегчению симптомов, коррекции аномалий и пороков развития, угрожающих жизни больных. Назначаются ноотропные средства, препараты кальция и витамина Д, для уменьшения выраженности умственной отсталости рекомендуют специализированную психологическую помощь. В ряде случаев при синдроме Рубинштейна-Тейби применяют хирургические методы лечения – для устранения пороков сердца, аномалий развития прямой кишки и мочевыделительных путей, крипторхизма. Также может потребоваться лечение у офтальмолога, а при признаках развития злокачественного новообразования – консультация и лечение у онколога.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 30 сентября 2013;
проверки требуют 11 правок.
Синдром Рубинштейна — Тейби (синоним: синдром широкого I пальца кистей и стоп, специфического лица и умственной отсталости) впервые описан в 1963 г. J. Rubinstein и Н. Taybi[2].
Популяционная частота — 1:25 000 — 30000, соотношение полов — 1:1.
Тип наследования аутосомно-доминантный (ген CREBBP в локусе 16p13.3), но в большинстве случаев мутация возникает спонтанно, то есть не наследуется от родителей[3][4].
Дифференциальный диагноз: брахидактилия, тип D, акроцефалосиндактилия, тип VI.
Минимальные диагностические признаки: прогрессирующая умственная отсталость, широкие терминальные фаланги первых пальцев кистей и стоп, характерное лицо, отставание роста и костного возраста, микроцефалия, крипторхизм.
Клиническая характеристика[править | править код]
Основными признаками синдрома являются отставание психофизического развития, дисплазия лица, аномалии развития пальцев. У 94 % больных рост ниже среднего. Костный возраст значительно отстаёт от паспортного (94 %). Отмечается олигофрения (100 %) (в большинстве случаев — тяжёлая умственная отсталость, задержка моторного и речевого развития). Характерна чрезмерная отвлекаемость, плохая концентрация внимания, трудность овладения речью[5].
Комплекс черепно-лицевых дисморфий включает брахицефалию, микроцефалию, большой и поздно закрывающийся родничок, выступающий лоб с низким ростом волос, приподнятые дугообразные брови, антимонголоидный разрез глаз (93 %), широкую переносицу, эпикант (62 %), длинные ресницы, птоз, широкую спинку носа (71 %), загнутый книзу кончик носа (клювовидный нос) (90 %), гипоплазию крыльев носа (72 %), умеренную ретрогнатию, гримасу, напоминающую улыбку, высокое арковидное небо (93 %).
Со стороны глаз отмечаются косоглазие (79 %) и аномалии рефракции (58 %). Ушные раковины деформированы, уменьшены или увеличены (74 %).
Аномалии пальцев заключаются в расширении, укорочении и уплощении ногтевых фаланг первых пальцев кистей и стоп (100 %), иногда терминальных фаланг других пальцев кисти (60 %), вальгусной деформации межфаланговых суставов, удвоении ногтевой фаланги первых пальцев стоп (30 %), реже — проксимальной фаланги, в ряде случаев — полидактилии стоп, частичной синдактилии кистей и стоп.
Отмечаются также лордоз, кифоз, сколиоз, аномалии грудины и рёбер, уплощение крыльев тазовых костей.
В половине случаев встречаются гирсутизм, ярко-красный невус на коже лба, затылка, боковой поверхности шеи. Часты изменения дерматоглифических показателей.
Пороки развития внутренних органов включают незаращение артериального протока и дефекты перегородок сердца, одностороннюю аплазию почек, удвоение почек, гидронефроз, расширение мочеточников или их стеноз, дивертикул мочевого пузыря, крипторхизм (в 79 % случаев), нарушение лобуляции легких, агенезию или аплазию мозолистого тела (обнаруживаемую при МРТ-исследовании).
Примечания[править | править код]
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ RUBINSTEIN J. H., TAYBI H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. (англ.) // American journal of diseases of children (1960). — 1963. — Vol. 105. — P. 588—608. — PMID 13983033.
- ↑ Wójcik C., Volz K., Ranola M., Kitch K., Karim T., O’Neil J., Smith J., Torres-Martinez W. Rubinstein-Taybi syndrome associated with Chiari type I malformation caused by a large 16p13.3 microdeletion: a contiguous gene syndrome? (англ.) // American journal of medical genetics. Part A. — 2010. — Vol. 152A, no. 2. — P. 479—483. — doi:10.1002/ajmg.a.33303. — PMID 20101707.
- ↑ Petrij F., Giles R. H., Dauwerse H. G., Saris J. J., Hennekam R. C., Masuno M., Tommerup N., van Ommen G. J., Goodman R. H., Peters D. J. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. (англ.) // Nature. — 1995. — Vol. 376, no. 6538. — P. 348—351. — doi:10.1038/376348a0. — PMID 7630403.
- ↑ Исаев Д. Н. Умственная отсталость у детей и подростков. Руководство. — СПб: Речь, 2003. — С. 58. — 397 с. — ISBN 5-9268-0212-1.
Литература[править | править код]
- «Наследственные синдромы и медико-генетическое консультирование», С. И. Козлов, Е. С. Еманова[прояснить]
Источник
Утратил силу — Архив
![]()
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Архив — Клинические протоколы МЗ РК — 2010 (Приказ №239)
Категории МКБ:
Синдромы врожденных аномалий, вовлекающих преимущественно конечности (Q87.2)
Общая информация
Краткое описание
Термин «синдром» используется для обозначения устойчивых сочетаний патологических симптомов, как уточненной этиологии, так и этиологически неясных. В клинической генетике и тератологии термин «синдром» имеет несколько значений. Он часто применяется как синоним «болезни», когда речь идет о единой нозологической форме с определенной этиологией. При этом синдромом чаще называют врожденные нарушения.
Протокол «Синдромы врожденных аномалий, вовлекающих преимущественно конечности»
Код по МКБ:
Q 87.2
Синдром:
— Холта-Орама
— Клиппеля-Треноне-Вебераа
— Отсуствия (недорозвития) ногтей, надколенника
— Рубинштейна-Тейби
— Сиреномиелии (сращение нижних конечностей)
— Тромбоцитопения с отсутствием лучевой кости (TAR-синдром)
— VATER
Q87.4 синдром Марфана
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники. Стандарты лечения
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID
Классификация
1. Умственная отсталость.
2. Легкая умственная отсталость.
3. Умеренная умственная отсталость.
4. Тяжелая умственная отсталость.
5. Глубокая умственная отсталость.
Диагностика
Диагностические критерии
Жалобы и анамнез: задержка в психоречевом развитии, снижение мышления, памяти, внимания, расторможенность, врожденные пороки развития конечностей.
Физикальное обследование
Синдром Холта-Орама (фенотипические проявления) — пороки развития верхних конечностей варьируют от изменений I пальца кисти (гипоплазия, трехфаланговый I палец) и гипоплазия лучевой кости (50%). Левая рука поражается чаще. У 50% больных I палец не противопоставлен остальным пальцам кисти. Из скелетных изменений наблюдаются также гипоплазия лопаток и ключиц, сколиоз, воронкообразная деформация грудины, клинодактилия, синдактилия, гипоплазия или аплазия других пальцев кисти и костей запястья (40%). В 85% случаев синдрома обнаруживаются врожденные пороки сердца: дефекты межпредсердной перегородки (наиболее часто), дефект межжелудочковой перегородки (50%), открытый артериальный проток, коарктация аорты, стеноз легочной артерии, пролапс митрального клапана. В редких случаях описывают гипертелоризм, отсутствие большой грудной мышцы, расщелину неба.
Синдром Клиппеля-Треноне-Вебера (синонимы: гемангиэктазия гипертрофическая; невус остеогипертрофический варикозный) — фенотипические проявления: типичным признаком синдрома является гипертрофия одной или нескольких конечностей, что связано с врожденным пороком развития артерио-венозной системы и лимфатических сосудов. Отмечается гиперплазия мягких тканей и кости пораженной конечности, трофические язвы и отеки. Гемангиомы бывают единичные или множественные, капиллярные, кавернозные, кистозные, различающиеся по размерам и локализации (любая часть тела, но чаще всего на голени, ягодицах, животе, нижней части туловища), преобладает одностороннее поражение. Описаны гиперпигментация кожи, синдактилия, полидактилия. Челюстно-лицевые аномалии включают асимметричную гипертрофию лица, микроцефалию, макроцефалию. Наблюдаются глаукома, катаракта, колобома.
Синдром Рубинштейна-Тейби (фенотипические проявления), синонимы: синдром широкого I пальца кисти и стопы с лицевыми аномалиями. Клиническая картина характеризуются умственной отсталостью, задержкой роста и специфическими особенностями лица и тела, из которых основные — короткий и широкий I палец кисти и стопы, своеобразное лицо с длинным загнутым носом, антимонголоидным разрезом глаз, гипертелоризмом, недоразвитием верхней челюсти, брахимикроцефальной структурой черепа. Отмечается низкий рост волос на лбу, иногда линия роста волос спускается в центре углом. У больных, как правило, высокое небо, встречается его расщелина. Часто имеются синдактилия и полидактилия стоп, реже кистей, косолапость, врожденный вывих бедра. Задержка в росте в основном постнатальная.
Тромбоцитопения с отсутствием лучевой кости (TAR-синдром). Тромбоцитопения отмечается в 100% случаев и появляется в первые 4 мес. жизни, но с возрастом число тромбоцитов повышается до нормального уровня. Тромбоцитопенические кризы могут провоцироваться стрессом, инфекциями и хирургическими вмешательствами. Нарушена агрегация тромбоцитов и уменьшено время их жизни. Мегакарициты отсутствуют в 66% случаев, в 12% случаев их количество снижено. В 60-70% случаев заболевания отмечается лейкемоидная реакция на 1-м году жизни, особенно в период кровотечений; в это время нарастает тромбоцитопения и появляется гепатоспленомегалия. Основной скелетной аномалией является двустороннее отсутствие или гипоплазия лучевой кисти (100%), часто в сочетании с аномалиями кисти (ограничения разгибания, радиальная девиация, гипоплазия карпальных костей и фаланг при обязательной сохранности I пальца), укорочением и деформацией локтевой кости, отсутствием ее (в 20% случаев — двусторонним, 8% — односторонним), аномалией плеча (50%).
Синдром Марфана — для больных типичны высокий рост и длинные тонкие конечности, особенно дистальные отделы. Часто отмечаются сколиоз (60%), кифоз, воронкообразная или килевидная грудная клетка, долихоцефалия, узкое лицо, высокое дугообразное небо, плоскостопие. Поражения глаз включают двусторонний подвывих хрусталика (75%), часто сочетающийся с иридодонезом (дрожание радужки). Отмечаются высокая степень миопии, отслойка сетчатки, мегалокорнеа и голубые склеры.
Ассоциация VATER — основные проявления включают аномалии позвоночника (полупозвонки, кифосколиоз, менингоцеле) (82%), дефекты перегородки и другие пороки сердца (50%), атрезию ануса (70%), трахеопищеводный свищ с атрезией пищевода (78%), дисплазию лучевой кости, преаксиальную полидактилию и синдактилию (65%), аномалию почек.
Лабораторные исследования: общий анализ крови и мочи.
Инструментальные исследования:
1. Электроэнцефалография (ЭЭГ): на ЭЭГ задержка формирования возрастной корковой ритмики, диффузные изменения электрогенеза.
2. Электромиография (ЭМГ).
3. Компьютерная томография головного мозга (КТ).
Показания для консультации специалистов:
— логопед;
— психолог;
— ЛОР-сурдолог;
— окулист;
— кардиолог;
— генетик;
— педиатр;
— ортопед;
— протезист;
— врач ЛФК;
— физиотерапевт.
Минимум обследования при направлении в стационар:
1. Общий анализ крови.
2. Общий анализ мочи.
3. АЛТ.
4. АСТ.
5. Кал на яйца глист.
Основные диагностические мероприятия:
1. Общий анализ крови.
2. Общий анализ мочи.
3. ЭЭГ.
4. Исследование слуха.
5. КТ головного мозга.
6. Логопед.
7. Психолог.
8. Окулист.
9. Кариотип.
10. Генетик.
11. ЭКГ.
12. Кардиолог.
13. УЗИ органов брюшной полости.
Дополнительные диагностические мероприятия:
1. ИФА на токсоплазмоз.
2. ИФА на цитомегаловирус.
3. Анализ мочи на обменные нарушения.
Дифференциальный диагноз
Признак Синдром | Скелетные аномалии | Глазная патология | Характерный признак |
Холта-Орама | Пороки развития верхних конечностей варьируют от изменений I пальца кисти (гипоплазия, трехфаланговый I палец) и гипоплазия лучевой кости (50%) дофокомелии. Левая рука поражается чаще. У 50% больных I палец не противопоставлен остальным пальцам кисти. Из скелетных изменений наблюдаются также гипоплазия лопаток и ключиц, сколиоз, воронкообразная деформация грудины, клинодактилия, синдактилия, гипоплазия или аплазия других пальцев кисти и костей запястья. | Гипертелоризм. | Врожденные пороки сердца: дефекты межпредсердной перегородки (наиболее часто), дефект межжелудочковой перегородки (50%), открытый артериальный проток, коарктация аорты, стеноз легочной артерии, пролапс митрального клапана. |
Клиппеля-Треноне-Вебера | Гипертрофия одной или нескольких конечностей, что связано с врожденным пороком развития артерио-венозной системы и лимфатических сосудов. | Глаукома, катаракта, колобома | Гемангиомы бывают единичные или множественные, капиллярные, кавернозные, кистозные. Различаются по размерам и локализации на любой части тела, но чаще всего на голени, ягодицах, животе, нижней части туловища. |
Рубинштейна-Тейби | Синдактилия и полидактилия стоп, реже кистей, косолапость, врожденный вывих бедра, задержка в росте в основном постнатальная. | Антимонголоидный разрез глаз, гипертелоризмом. | Своеобразное лицо с длинным загнутым носом, антимонголоидным разрезом глаз, гипертелоризмом, недоразвитием верхней челюсти, брахимикроцефальной структурой черепа. |
Марфана | Высокий рост и длинные тонкие конечности, особенно дистальные отделы, часто отмечаются сколиоз (60%), кифоз, воронкообразная или килевидная грудная клетка, плоскостопие. | Двусторонний подвывих хрусталика (75%), часто сочетающийся с иридодонезом (дрожание радужки). Отмечаются высокая степень миопии, отслойка сетчатки, мегалокорнеа и голубые склеры. | Арахнодактилия, высокий рост, гиперподвижность суставов, подвывих хрусталика, аневризма аорты. |
Лечение
Тактика лечения: симптоматическое лечение.
Цели лечения:
— активизация психического развития;
— пополнение пассивного и активного словарного запаса;
— коррекция поведения;
— повышение эмоционального тонуса, настроения ребенка;
— обучение навыкам самообслуживания;
— социальная адаптация.
Немедикаментозное лечение:
— индивидуальные занятия с логопедом;
— занятия с психологом;
— кондуктивная педагогика;
— ЛФК;
— массаж;
— физиолечение.
Медикаментозное лечение
1. Широко используют в последнее время препараты ноотропного ряда — нейропротекторы, с целью улучшения обменных процессов в головном мозгу. Большинство ноотропных препаратов в связи с их психостимулирующим действием назначают в первую половину дня. Продолжительность курсов лечения ноотропами составляет от одного до двух-трех месяцев:
— церебролизин, ампулы 1 мл в/м;
— пирацетам, ампулы 5 мл 20%;
— гинкго-билоба (танакан), таблетки 40 мг;
— пиритинол гидрохлорид (энцефабол), драже 100 мг, суспензия — 5 мл содержит 80,5 мг пиритинола (соотв. 100 мг пиритинола гидрохлорида). Энцефабол — минимум противопоказаний, разрешен к применению с первого года жизни. Дозирование суспензии (с содержанием в 1 мл 20 мг энцефабола) детям 3-5 лет — суточная доза 200-300 мг (12-15 мг массы тела) назначают в 2 приема — утром (после завтрака) и днем (после дневного сна и полдника). Продолжительность курса 6-12 недель, целесообразен длительный прием, при котором повышается работоспособность и способность к обучению, улучшаются высшие психические функции;
— актовегин, ампулы 2 мл 80 мг, драже-форте 200 мг активного вещества. Нейрометаболический препарат, содержащий исключительно физиологические компоненты. Детям назначается в драже-форте, прием до еды по ½ -1 драже 2-3 раза в день (в зависимости от возраста и выраженности симптомов заболевания) до 17 часов. Продолжительность терапии 1-2 месяца;
— инстенон, таблетки (1 таблетка содержит 50 мг этамивана, 20 мг гексобендина, 60 мг этофиллина). Многокомпонентный нейрометаболический препарат. Суточная доза составляет 1,5-2 таблетки, назначается в 2 приема (утром и днем) после еды. Для исключения побочных эффектов рекомендуется постепенное наращивание дозы в течение 5-8 дней. Продолжительность лечения 4-6 недель.
2. Ангиопротекторы с целью улучшения мозгового кровообращения: винпоцетин, циннаризин.
3. Витамины группы В: В1, В6, В12, нейромультивинт — специальный комплекс витаминов группы В с направленным нейротропным действием, неуробекс, фолиевая кислота, аевит.
4. Седативная терапия по показаниям: ноофен, ново-пассит.
5. Корректоры поведения: сонапакс, хлорпротиксен.
Профилактические мероприятия:
— профилактика травматизма;
— профилактика вирусных и бактериальных инфекций.
Дальнейшее ведение: регулярные занятия с логопедом, дефектологом, психологом, социальная адаптация ребенка, оформление в специализированный детский сад, прохождение медико-педагогической комиссии для решения вопроса об обучении ребенка.
Основные медикаменты:
1. Актовегин, ампулы 2 мл, 80 мг
2. Винпоцетин, таблетки 5 мг
3. Пирацетам, ампулы 5 мл, 20%
4. Пирацетам, таблетки 0,2 и 0,4
5. Пиридоксин гидрохлорид, ампулы, 1 мл 5%
6. Тиамин хлорид, ампулы 5%, 1 мл
7. Фолиевая кислота, таблетки 0,001
8. Церебролизин, ампулы 1 мл
9. Цианокобаламин, ампулы 1 мл 200 мкг и 500 мкг
Дополнительные медикаменты:
1. Аевит, капсулы
2. Глицин, таблетки 0,1
3. Гопантеновая кислота (пантокальцин), таблетки 0,25
4. Инстенон, таблетки
5. Нейромультивит, таблетки
6. Неуробекс, таблетки
7. Ново-пассит, таблетки, раствор
8. Ноофен, таблетки 0,25
9. Пиритинол, драже 0,1, суспензия
10. Сонапакс, таблетки 10 мг
11. Хлорпритиксен 15 мг
12. Циннаризин, таблетки 25 мг
Индикаторы эффективности лечения:
— улучшение внимания, памяти, работоспособности;
— пополнение пассивного и активного запаса слов;
— повышение эмоционального и психического тонуса.
Госпитализация
Показания к госпитализации (плановая): задержка психоречевого и моторного развития, различные врожденные пороки развития конечностей.
Информация
Источники и литература
- Протоколы диагностики и лечения заболеваний МЗ РК (Приказ №239 от 07.04.2010)
- Перинатальная патология» под редакцией М.Я. Студеникина. Москва 1984
Ю.А. Якунин. Болезни нервной системы у новорожденных и детей раннего возраста. Москва 1979
Справочник детского психиатра и невропатолога под редакцией Л.А. Булаховой. Киев 1997
Л.О. Бадалян. Детская неврология. Москва 1998
Г.С. Маринчева, В.И. Гаврилов. Умственная отсталость при наследственных болезнях. Москва 1988
С.К. Козлова, Е. Семанова. Наследственные синдромы и медико-генетическое консультирование. Ленинград 1987
- Перинатальная патология» под редакцией М.Я. Студеникина. Москва 1984
Информация
Список разработчиков:
№ | Разработчик | Место работы | Должность |
1. | Кадыржанова Галия Баекеновна | РДКБ «Аксай», психоневрологическое отделение №3 | Заведующая отделением |
2. | Серова Татьяна Константиновна | РДКБ «Аксай», психоневрологическое отделение №1 | Заведующая отделением |
3. | Мухамбетова Гульнара Амерзаевна | Кафедра нервных болезней, КазНМУ | Ассистент, кандидат медицинских наук |
4. | Балбаева Айым Сергазиевна | РДКБ «Аксай», психоневрологическое отделение №3 | Врач-невропатолог |