Синдром птицеголовая карликовость или синдром секкеля

Синдром Секкеля является унаследованной формой врожденной карликовости , а это значит, что младенец начинает очень мало и не может нормально расти после рождения. В то время как люди с синдромом Секкеля обычно будут пропорциональны по масштабу, у них будет отчетливо малый размер головы. Также распространена умственная отсталость.

Несмотря на множество физических и умственных проблем, с которыми сталкивается человек с синдромом Секкеля, многие из них, как известно, хорошо живут более 50 лет.
Причины синдрома Секкеля
Синдром Секкеля является наследственным расстройством, связанным с генетическими мутациями на одной из трех разных хромосом. Считается, что это бывает крайне редко, с немногим более 100 случаев, зарегистрированных с 1960 года. Многие дети, получившие диагноз синдрома Секкеля, родились у родителей, которые тесно связаны (родственники), например, брак с кузенами или братьями и сестрами.
Синдром Секкеля является рецессивным генетическим заболеванием, он возникает только тогда, когда ребенок наследует один и тот же ненормальный ген от каждого родителя. Если ребенок получает один нормальный ген и один ненормальный ген, ребенок будет носителем синдрома, но обычно не проявляет симптомов.
Если оба родителя имеют одинаковую хромосомную мутацию для синдрома Секкеля, их риск иметь ребенка с синдромом Секкеля составляет 25%, а риск наличия носителя — 50%.
Характеристики синдрома Секкеля
Синдром Секкеля характеризуется аномально медленным развитием плода и низким весом при рождении.
После рождения у ребенка будет медленный рост и созревание костей, что приводит к короткому, но пропорциональному росту (в отличие от ахондроплазии). Лица с синдромом Секкеля обладают различными физическими и характеристиками развития, в том числе:
Очень маленький размер и вес при рождении
- Чрезвычайно малый, пропорциональный рост
- Аномально небольшой размер головы (микроцефалия)
- Клювообразный выступ носа
- Узкое лицо
- Нарушенные уши
- Необычно маленькая челюсть (микрогнатия)
- Умственная отсталость, часто тяжелая с IQ менее 50
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, повреждение зубов и другие деформации кости. Расстройства крови, такие как анемия (низкие эритроциты), панцитопения (недостаточно клеток крови) или острый миелоидный лейкоз.
В некоторых случаях семенники у мужчин не спускаются в мошонку, а женщины могут иметь аномально увеличенный клитор. Кроме того, у людей с синдромом Секкеля могут быть чрезмерные волосы на теле и единая глубокая складка на ладонях (известная как складки обезьян).
Диагностика синдрома Секкеля
Диагноз синдрома Секкеля основан почти исключительно на физических симптомах. Возможно, потребуется рентгеновское изображение и другие инструменты (МРТ, КТ), чтобы отличить его от других подобных условий. В настоящее время нет лабораторных или генетических тестов, специфичных для синдрома Секкеля. В некоторых случаях окончательный диагноз не может быть сделан до тех пор, пока ребенок не станет старше и не появятся характерные симптомы.
Лечение синдрома Секкеля
Лечение синдрома Секкеля сосредоточено на любой медицинской проблеме, которая может возникнуть, особенно при нарушениях крови и структурных деформациях. У людей с психическими расстройствами и их семьям должна быть предоставлена соответствующая социальная поддержка и консультационные услуги.
Источник
Синдром Секкеля является одним из врожденных заболеваний которые влияют на развитие людей от стадии беременности до рождения и имеют последствия для внешнего вида и основных биологических функций, а также для психических процессов.
Хотя идентифицирующий элемент, который наиболее очевиден, низкий рост или карликовость и, в большинстве случаев, форма носа, за этой клинической картиной есть много других необычных проявлений, которые могут серьезно подорвать качество жизни человека, если не будут приняты соответствующие меры.
В этой статье мы увидим, что они симптомы и известные причины синдрома Секкеля, а также его лечение .
- Связанная статья: «Карликовость: причины, симптомы и связанные расстройства»
Что такое синдром Секкеля?
То, что мы знаем как синдром Секкеля набор физических и психических изменений, которые появляются до рождения и у них есть генетические причины в их основе.
Это редкое заболевание, для которого характерно замедление развития плода, так что его уровень развития «задерживается» во многих аспектах.
симптомы
Основные признаки и симптомы, связанные с синдромом Секкеля, следующие.
1. Микроцефалия
Это один из наиболее характерных признаков синдрома Секкеля и состоит из недостаточное развитие свода черепа , что делает голову маленькой и, следовательно, мозг имеет меньше места для роста. Это важно, учитывая, что эта часть нервной системы должна быстро расширяться во время этой фазы жизни.
- Статья по теме: «Микроцефалия: симптомы, характеристики и лечение»
2. Умственная отсталость
Это одно из последствий уменьшенного размера черепа по отношению к остальной части тела. Пороки развития мозга это следствие этого ограничения пространства часто вызывает более ограниченное когнитивное развитие, чем обычно, хотя это не оценивается во время рождения.
- Может быть, вы заинтересованы: «Типы умственных недостатков (и характеристики)»
3. Птичий профиль
Это имя, под которым называется лицо, которое часто встречается у детей с синдромом Секкеля. Это связано с уменьшенным размером головы, сравнительно большими глазами и очень выраженный носовой мост, который дает ощущение «пика» .
4. Маленький рост или карликовость
В целом, люди с синдромом Секкеля меньше, чем ожидается для их возрастной группы. Это также влияет на пропорции, так как конечности маленькие s пропорционально остальной части тела.
С другой стороны, вследствие медленного развития созревания конфигурация кости также слабо развита, что может привести к появлению некоторых скелетных нарушений.
5. Другие признаки и симптомы
Есть и другие общие признаки и симптомы синдрома Секкеля, среди которых распространены следующие:
- Диспластические уши : развитие ушей также недостаточно, так что их дизайн не так сложен, как это обычно бывает у других людей.
- Дисплазия зубов Зубы слабо развиты и их распределение нарушено, что иногда мешает хорошо закрыть рот.
- косоглазие : глаза отклонены, так что они не указывают наружу параллельно.
- Дефекты на вкус : часть неба представляет изменения, такие как наличие отверстий или свод, который является слишком явным и узким.
диагностика
Синдром Секкеля можно ожидать на стадии развития плода с помощью ультразвука (обзор пороков развития и развития костей), хотя диагноз не ставится до тех пор, пока признаки и симптомы не успеют проявить себя, что происходит на первой стадии детства, но не в недели сразу после рождения.
причины
В настоящее время причины синдрома Секкеля плохо изучены. Тем не менее, известно, что это изменение на основе генетических триггеров аутосомно-рецессивного типа Это означает, что дефектная версия определенного гена должна присутствовать как у отца, так и у матери, чтобы потомство выражало симптомы.
С другой стороны, нет ни одного гена, который бы вызывал симптомы синдрома Секкеля, но в настоящее время известны три генетических изменения, связанные с этим заболеванием. В свою очередь, это различное происхождение уступает различным типам синдрома Секкеля, связанным с хромосомами 3, 14 и 18.
Лечение синдрома Секкеля
Синдром Секкеля не имеет известного лечения, так как он имеет генетическое происхождение и его эффекты начинают замечаться с момента образования плода. Тем не менее, есть несколько стратегий, которые могут помочь смягчить негативные последствия, которые вызывают симптомы .
В этом смысле междисциплинарный подход и тренировка навыков, направленных на предотвращение проблем, связанных с конкретными осложнениями например, проблемы с масикаром или дыханием, а также терапия, направленная на изучение норм поведения и отношений с другими. Подобные инициативы должны быть адаптированы к уровню интеллектуального развития каждого пациента.
НА Пабло Уэртас. В авангарде европейской молекулярной биологии (June 2020).
Источник
Синдром ICF (акроним от англ. Immune deficiency
— иммунодефицит, Centromeric instability- нестабильность
центромерного гетерохроматина, Facial dysmorphism
— лицевые аномалии). Аутосомно-рецессивное
заболевание (MIM242860), основными клиническими
признаками которого являются лицевые
аномалии, дефицит IgA и Т-клеток, инфекции
верхних дыхательных путей и склонность
к злокачественным новообразованиям.
Цитогенетически характеризуется нестабильностью
центромерных гетерохроматиновых районов
хромосом 1, 9,16 и реже — 2 и 10, с образованием
множественных ветвящихся структур. Недавно
было обнаружено, что в основе этиологии
синдрома лежит мутация гена, картированного
в сегменте 20q12, который кодирует ДНК-метилтрансферазу
ЗВ типа (DNMT3B). Мутация этого гена приводит
к нарушению метилирования классической
сателлитной ДНК, локализованной в центромерных
гетерохроматических районах, что и приводит
к нестабильности этих участков генома,
выявляемой на хромосомном уровне.
Порокератоз Мибелли. Заболевание с
аутосомно-доминантным типом наследования
(MIM 175800). Проявляется в видекожной сыпи
с кратерообразно углубленными участками
атрофии и центробежно распространяющимися
кожнымилоскутами, окруженными узкими
роговыми гребнями. Начинается в среднем
возрасте и может приводить к раку кожи,
особенно на конечностях. Обнаруживается
высокая частота хромосомных нарушений
в фибробластах кожи. Часто повреждается
сегмент Зр14-р12.
Птицеголовая карликовость
или синдром Секеля. Заболевание
с аутосомно-рецессивным типом наследования
(MIM210600). Характеризуется низким весом
при рождении, карликовостью, маленькой
головой, клювовидным носом, большими
глазами, узким лицом, умственной отсталостью,
маленьким мозгом, панцитопенией и повышенной
частотой хромосомных аберраций.
Рассмотренные выше заболевания
проявляются повышенной частотой хромосомных
аберраций, которые, как правило, не связаны
с повреждением строго определенных хромосом
и хромосомных сегментов (за исключением
синдромов Луи-Бар и Ниймегена). Однако
в хромосомах человека имеется достаточно
большое количество сайт-специфическихломких
участков, экспрессия которых существенно
зависит от условий культивирования клеток
и молекулярная природакоторых только
сейчас начинает проясняться.
Синдром
ломкой Х-хромосомы
Как
правило, разрывы хромосом или пробелы
хроматид, возникающие с повышенной частотой
в тех или иных конкретных хромосомных
сегментах (так называемые ломкие участки
или фрагильные сайты хромосом), не связаны
с какими-либо заболеваниями. Однако из
этого правила есть исключение. В1969 году
американский исследователь Lubs у больных
ссиндромом, сопровождающимся умственной
отсталостью, обнаружил наличие специфического
цитогенетического маркера — в дистальной
части длинного плеча Х-хромосомы в сегменте
Xq27.3 в отдельных клетках обнаруживался
разрывили пробел хроматид. Позднее было
показано, что первое клиническое описание
семьи с синдромом, в котором умственная
отсталость является ведущим клиническим
признаком, был описан еще в 1943 году английскими
врачами Purdon Martin и Julia Bell. Синдром Мартина-Белл
или синдром ломкой Х-хромосомы характеризуется
ломкой (фрагильной) Х-хромосомой в сегменте
Xq27.3, которая выявляется в специальных
условиях культивирования клеток в среде
с дефицитомфолиевой кислоты. Фрагильный
сайт при этом синдроме получил обозначение
FRAXA. Основными диагностическимипризнаками
заболевания являются: умственная отсталость,
прогнатизм, широкое лицо с чертами акромегалии,
большие оттопыренные уши, макроорхидизм
в постпубертатном периоде, аутизм, гиперкинезы,
плохая концентрация внимания, дефекты
речи, более выраженные у детей. Отмечаются
также аномалии соединительной ткани
с гиперрастяжимостьюсуставов и пролапсом
митрального клапана. Относительно полный
спектр клинических признаков имеют только
60% мужчин с фрагильной Х-хромосомой, 10%
больных не имеют лицевых аномалий, 10%
имеют только умственную отсталость без
других признаков, а 30% больных не имеют
макроорхидизма.
Синдром фрагильной
Х-хромосомы интересен своим необычным
наследованием и высокой популяционной
частотой (1 на 1500-3000). Необычность наследования
состоит в том, что только 80% мужчин, носителей
мутантного гена, имеют клинические и
клинические признаки заболевания, а остальные
20% как клинически, так и цитогенетически
нормальны, хотя после передачи мутации
своим дочерям могут иметь пораженных
внуков. Этих мужчин называют трансмиттерами,
т.е. передатчиками неэкспрессированного
мутантного гена, который становится экспрессируемым
в последующих поколениях. Кроме того,
имеется 2 типа женщин — гетерозиготных
носителей мутантного гена: а) дочери мужчин-трансмиттеров,
неимеющих симптомов заболевания, у которых
не выявляется фрагильная X хромосома;
б) внучки нормальных мужчин-трансмиттеров
и сестры пораженных мужчин, которые обнаруживают
клинические признаки заболевания в 35%
случаев. Таким образом, мутация гена при
синдроме Мартина-Белл существует в двух
формах, отличающихся по своей пенетрантности:
первая форма-фенотипически не проявляющаяся
премутация, которая переходит в полную
мутациювторая форма при прохождении
через женский мейоз. Обнаружена четкая
зависимость развития умственной отсталости
отположения индивида в родословной. При
этом хорошо прослеживается явление антиципации
— более тяжелого проявления заболевания
в последующих поколениях.
Молекулярный механизм мутации
стал понятен в 1991 году, когда был охарактеризован
ген, ответственный за развитиеданного
заболевания. Ген получил название FMR1
(англ. — Fragile site Mental Retardation 1 — ломкий участок
хромосомы, связанный с развитием умственной
отсталости 1 типа). Было установлено, что
в основе клинических проявлений ицитогенетической
нестабильности в локусе Xq 27.3 лежит многократное
увеличение в первом экзоне гена FMR-1 простого
тринуклеотидного повтора CGG. У нормальных
людей число этих повторов в Х-хромосоме
колеблется от 5 до 52, а у больных их число
составляет 200 и более. Такое явление резкого,
скачкообразного изменения числа CGG-повторов
у больных получило название экспансии
числа тринуклеотидных повторов.
Показано,
что экспансия CGG-повторов существенно
зависит от пола потомка, она заметно
увеличена при передачи мутации от матери
к сыну. Важно отметить, что экспансия
нуклеотидных повторов является постзиготическим
событием и возникает на очень ранних
стадиях эмбриогенеза Кроме ломкого сайта
FRAXA в дистальнойчасти длинного плеча
Х-хромосомы в настоящее время идентифицировано
еще 3 других ломких участка — FRAXE, FRAXF и
RFAXD. Первые два связаны с аномалиями развития,
а ломкий участок RFAXD, расположенный недалеко
от сайта FRAXA, выявляется с частотой 1-2%
у нормальных лиц и может приводить к ложно-позитивной
диагностике синдрома.
Источник
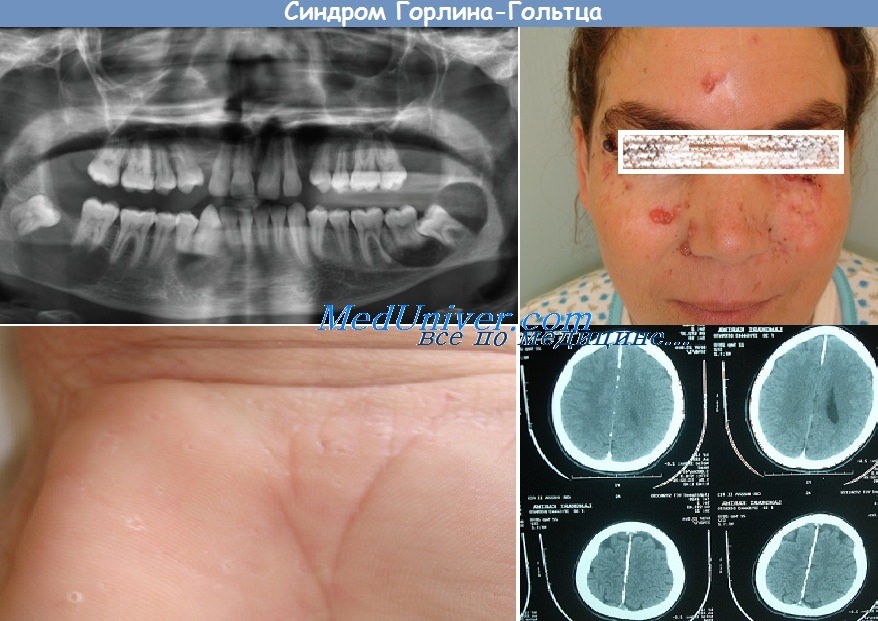
Синдром Горлина—Гольтца. Диагностика и лечение синдрома Горлина-Гольтца.Синдром Горлина—Гольтца (син.: синдром невоидной базальноклеточной карциномы, синдром базальноклеточных невусов) — генетически детерминированный полиорганный синдром, наследуемый по аутосомно-доминантному типу с высокой пенетрантностью и различной экспрессивностью. Мутантный ген локализуется в хромосоме 9 на участке q—22.3-q31. Основное проявление синдрома — множественные базалиомы, которые ассоциируются с разнообразными пороками развития скелета, глаз, нервной, эндокринной систем и других органов и тканей, а также с опухолями различной локализации. Заболевание обусловлено мутациями гена PTSH в хромосоме 9 и ассоциируется с антигенами Н1А-А10, В14. Почти все изменения врожденные. Базалиомы также могут быть врожденными, хотя чаще появляются в позднем детском возрасте, как правило они развиваются у лиц не старше 35 лет. Обычно это множественные базалиомы, располагающиеся симметрично и билатерально как на открытых, так и на закрытых участках кожи. Преимущественно поражаются лицо, шея, туловище и конечности. Количество базалиом может достигать нескольких сотен. Вначале процесс представлен поверхностными базалиомами диаметром от 1 до 3 см, развивающимися медленно и практически не меняющимися по величине до наступления 2-го или 3-го десятилетия жизни. Затем отдельные новообразования могут быстро увеличиваться до 5-10 см. изъязвляться, под влиянием неблагоприятных внешних факторов среди них появляются кистоз-ные, язвенные формы, а также метатипический рак кожи. Точечные пигментированные углубления на ладонях и подошвах нередко возникают раньше других кожных проявлений синдрома и встречаются в 70-80% случаев, на их дне почти всегда имеются телеангиэктазии. Количество элементов — до нескольких сотен, больше всего их на боковых поверхностях ладоней, подошв и пальцев кистей. Они возникают из-за преждевременного слущивания рогового слоя эпидермиса. Изредка из углублений в области ладоней и подошв могут развиваться опухоли. Синдром Горлина—Гольтца также может ассоциироваться с другими поражениями кожи: эпидермальными кистами, милиумом, фибромами, липомами, пигментными невусами, веснушками, ладонно-подошвенным гиперкератозом, комедонами . Лишь у отдельных больных с синдромом Горлина—Гольтца отсутствуют кожные опухоли. Среди различных костных аномалий, которые имеют врожденный характер и выявляются у 75-90% больных с этим синдромом, наиболее характерны множественные одонтогенные кисты верхней и нижней челюстей которые при нагноении приводят к отечности и тупой боли и могут вскрываться в полость рта. Описываются также кифоз, сколиоз, расщепление ребер, воронкообразная грудная клетка, укорочение IV пястных костей, синостозы, прогнатия, парадонтоз, неправильное расположение зубов, готическое нёбо, срединный носовой синус, широкие носовые ходы, истинный гипертелоризм, выступающие лобные бугры, дизостозы костей лицевого черепа, субкорнеальные кистозные изменения длинных трубчатых и плоских костей. Аномалии глаз встречаются у 26% больных и проявляются врожденной слепотой, гипертелоризмом, дистопией внутренних углов глаза, катарактой, глаукомой, колобомой, стробизмом.
Поражения центральной нервной системы отмечаются у 10-42% больных и проявляются гидроцефалией, микроцефалией, недоразвитием мозолистого тела головного мозга, пластинчатым обызвествлением серпа большого мозга, кальцификацией твердой и мягкой мозговых оболочек, эпилепсией, деменцией, задержкой психического развития. Патология со стороны эндокринной и половой систем (крипторхизм, гипогонадизм, бесплодие, двурогая матка, фиброматоз яичников, акромегалия, тиреоидит) наблюдается у 17% больных. Синдрому Горлина— Гольтца могут сопутствовать и другие аномалии развития: врожденное отсутствие почки и мочеточника, анемия Фанкони и др. Синдром Горлина— Гольтца ассоциируется с такими опухолями, как медуллобластома, фиброматоз яичников, фибросаркомы челюстей, фибромы, тератомы и цистаденомы яичников, лейомиомы, нейрофиброматоз I типа, болезнь Ходжкина, ретинобластома, менингиома, рабдомиома, множественные миомы матки, семинома, рак матки, ренинсекретирующая опухоль яичника, опухоль мозжечка, менингиома. В частности, медуллобластома (мутации в гене 9q.31) чаще поражает мальчиков в возрасте до 2 лет, в связи с чем нередко возникает до появления кожных опухолей. Другие опухоли при синдроме Горлина—Гольтца, в отличие от первично-множественных базалиом, протекают более агрессивно и резистентно по отношению к проводимой терапии, часто рецидивируют и даже могут метастазировать. Диагноз синдрома Горлина— Гольтца устанавливается на основании совокупности клинико-гистологических данных и результатов обследования стоматологом, челюстно-лицевым хирургом, невропатологом, окулистом, эндокринологом, гинекологом; обязательна рентгенография плоских костей, а для выявления пластинчатого обызвествления серпа большого мозга — рентгенологическое исследование черепа. Дифференциальный диагноз проводят с первично-множественными базалиомами. Течение синдрома Горлина— Гольтца сопровождается появлением новых опухолей на протяжении всей жизни. Прогноз лучше, чем при медуллобластоме, возникающей независимо от синдрома Горлина—Гольтца. Опухоли, расположенные в центральной части лица, на ушных раковинах или в их окружности, подлежат иссечению по методу Mohs — с микроскопией замороженных горизонтальных срезов для определения объема операции. Мелкие опухоли на туловище и конечностях удаляют путем электрокоагуляции или проводят комбинированное лечение этретинатом и флуороурацилом (5% крем) в течение 25-30 сут. В профилактических целях используют этретинат.
— Также рекомендуем «Нейрофиброматоз. Причина и признаки нейрофиброматоза.» Оглавление темы «Генетически обусловленные кожные заболевания.»: |
Источник