Синдром при котором длинные конечности

Врожденные аномалии, особенно те, которые передаются по аутосомно-доминантному типу, впечатляют своим разнообразием. Одним из таких недугов является синдром Марфана. Для данной болезни характерно наличие целого комплекса выраженных клинических симптомов. Лечение недуга специфическое, а потому требует детального рассмотрения.
Источник: https://vseolady.ru/sindrom-marfana.html
Что это за недуг
Синдром Марфана – что это такое? Это генетическое заболевание, возникающее на фоне мутации гена, отвечающего за синтез фибриллина. Этот белок является важной структурной единицей соединительной ткани. Именно благодаря ему, она обладает известной способностью к растяжению и сокращению, без появления разрывов.
Нехватка фибриллина – это прямой путь к патологии соединительной ткани. Волокна формируются неправильно, они не такие прочные, эластичные и упругие. Вследствие этого, ткань неспособна выдерживать традиционные для человека нагрузки. Она может быстро и в большом количестве разрываться, на месте повреждений появляются характерные рубцы – стрии. Растяжки на спине у подростков, в паху, под руками и прочих частях тела могут появляться именно из-за проблем с развитием соединительной ткани.
В 75% диагностированных случаях болезнь имела семейный тип наследования, и только в 25 процентах наблюдалась первичная мутация. Есть гипотеза, что недуг передается ребенку преимущественно от отца. Если у него или у его родственников был поставлен такой диагноз, вероятность того, что болезнь передастся ребенку, существенно повышается.
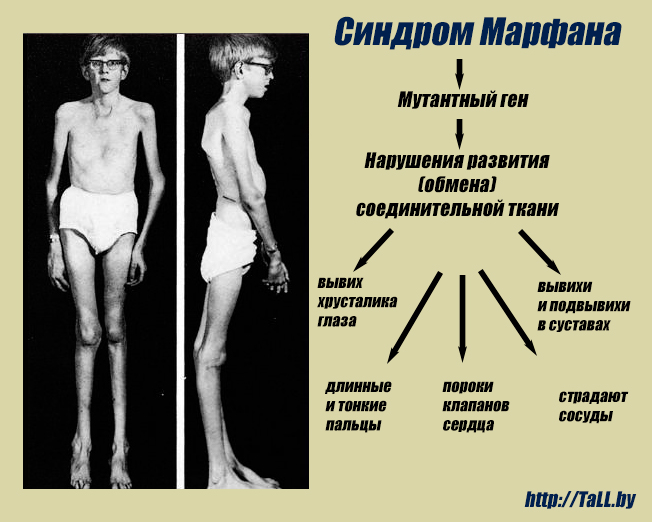
Синдром Марфана – генетическое заболевание, возникающее на фоне мутации гена, отвечающего за синтез фибриллинаОтличительной особенностью этого заболевания является то, что оно может поражать сразу несколько систем человеческого организма. Люди с синдромом Марфана выглядят соответствующим образом – у них, как правило, высокий рост, но маленькое туловище, из-за чего конечности (как нижние, так и верхние) выглядят непропорционально длинными. Пальцы тоже очень длинные, из-за чего их нередко называют паучьими, наблюдается вдавленная грудная клетка. Из-за натянутой кожи кажется, что больные намного моложе, чем есть на самом деле.
Синдром Марфана – что это за болезнь:
Форма заболеванияТяжесть протеканияХарактер течения
Выделяют две основных формы синдрома:
- Стертая – клинические проявления незначительные, и наблюдаются преимущественно в одной, максимум – в двух системах организма (например, сердечно-сосудистой и опорно-двигательного аппарата).
- Выраженная – симптоматика хорошо выражена в одной, двух, трех или больше системах. Также к такой форме относят тип недуга, когда симптоматика выражена слабо, но она затрагивает больше двух систем человеческого организма.
Данное заболевание может протекать в легкой, средней и тяжелой форме. Чем тяжелее течение, тем сильнее выражены симптомы, и тем больше поражение организма. В самых крайних случаях функциональность организма нарушена практически полностью, пациенту грозит летальный исход.Синдром может быть стабильным, то есть, таковым, при котором клинические проявления остаются примерно на одном уровне – не утихают, но и не усиливаются. При прогрессирующей форме негативные признаки заболевания постоянно усиливаются, постепенно ухудшая состояние больного.
Синдром Марфана – симптомы
Поскольку аутоимунный недуг затрагивают сразу несколько систем человеческого организма, нет ничего странного в том, что признаков очень много. Некоторые из них выражены лучше, другие – хуже. Ряд симптомов схож с проявлениями других заболеваний, например:
- гомоцистинурия;
- врожденная контрактурная арахнодактилия (синдром Билса);
- наследственная артроофтальмопатия (синдром Стиклера);
- MASS-синдром;
- синдромы Элерса-Данлоса, Лойса-Дитца, Шпринцена-Голдберга;
- семейная эктопия хрусталика.
Проявляющую симптоматику можно разделить на несколько групп, что поможет лучше понять ее особенности.
- Опорно-двигательный аппарат
Как уже говорилось выше, больные этим заболеванием имеют преимущественно высокий рост, короткое туловище и длинные конечности с такими же пальцами. Отмечается долихоцефалия – длинный и узкий лицевой скелет. Мышцы и подкожная клетчатка развиты крайне слабо. У некоторых больных присутствует нарушение прикуса и высокое, аркообразное небо.
Источник: https://vseolady.ru/sindrom-marfana.html
Помимо этого, признаки синдрома Марфана со стороны скелетной системы также включают в себя:
- килевидная или воронкообразная грудная клетка;
- плоскостопие;
- кифосколиотическая деформация позвоночника;
- нарушения функции суставов (гипермобильность);
- протрузия вертлужной впадины.
Симптомы нарушений функциональности опорно-двигательного аппарата являются одними из основных показателей для постановки правильного диагноза, поскольку они, как правило, хорошо выражены, и присутствуют у подавляющего количества пациентов.
- Мышечная система
К классическим проявлениям данного заболевания в данном случае можно отнести мышечную гипотонию, гипоплазию жировой ткани, практически полное отсутствие мускулатуры, а также небольшую массу тела. Это плохо для всех людей, но для беременных женщин – особенно, так как их кожа практически не растягивается, и под ней, по сути, ничего нет. Проблема в том, что подобные изменения характерны и для многих других недугов. Соответственно, здесь нужно либо дифференцировать диагноз, либо обращать внимание на другие симптомы.
- Сердечно-сосудистая система
Например, те, которые проявляются со стороны сердечно-сосудистой системы. Как правило, они доминируют, а изменения, затронувшие эту систему, чаще всего определяют исход заболевания. Преимущественно страдает сердце, однако патологии актуальны и для сосудов. Симптомов здесь может быть очень много, и практически все они несут определенную угрозу здоровью и даже жизни пациента:
- вегетативные расстройства;
- незначительные признаки гипертрофии миокарда левого желудочка и предсердия;
- проблемы с внутрижелудочковой проходимостью;
- поражение митрального клапана;
- патологическое удлинение и разрыв створочных хорд;
- нарушения ритма, тахикардия, фибрилляция;
- инфекционный эндокардит;
- прогрессирующее расширение восходящей части и клапанного кольца аорты;
- аневризмы.
На фоне проблем с сердцем и сосудами наблюдается и мышечная слабость. Человек чувствует себя постоянно разбитым, минимальные физические нагрузки даются ему с большим трудом. Данное заболевание может также привести к образованию врожденных пороков сердца – стеноза легочной артерии, коарктации аорты и т.д.
- Зрительная система
У многих больных наблюдаются проблемы и с органами зрения. В первую очередь, к традиционным симптомам можно отнести:
- миопия (более известна, как близорукость);
- эктопия (подвывих хрусталика);
- уплощение и увеличение размера роговицы;
- гипоплазия радужной оболочки – аниридия;
- расширение или сужение сосудов глаза;
- косоглазие;
- афакия;
- колобома.
Основная проблема в том, что негативные проявления со стороны зрительной системы наблюдаются уже с самого раннего возраста и, как правило, постоянно прогрессируют. Если не придавать этому факту должного значения, то это приведет к существенному ухудшению функции зрения, вплоть до слепоты.
- Нервная система
В первую очередь, это эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле, анизокория, пирамидные расстройства, асимметрия сухожильных рефлексов. Могут быть и другие проявления – это еще до сих пор изучается современной наукой.
Признаки синдромаДля данного заболевания характерны и другие проблемы с внутренними органами и тканями, например:
- спонтанный пневмоторакс;
- эмфизема легких;
- дыхательная недостаточность;
- атрофические стрии;
- паховые и бедренные грыжи, которые постоянно рецидивируют;
- вывихи и разрывы связок;
- эктопия почек;
- опущение мочевого пузыря и матки;
- варикозное расширение вен.
Диагностика синдрома Марфана и лечение
Диагностировать это заболевание можно с помощью семейного анамнеза, наблюдения за клиническими симптомами, физического осмотра пациента, а также на основе данных, собранных посредством соответствующих диагностических и лабораторных мероприятий:
- электрокардиограмма;
- ЭхоКГ;
- анализы крови;
- офтальмологический осмотр;
- генетические исследования;
- рентген;
- функциональная диагностика;
- компьютерная томография;
- магнитно-резонансная терапия;
- аортография.
Поскольку причины симптома Марфана изучены достаточно хорошо, главная задача врачей – это дифференцировать данное заболевание от других патологий. Это очень важно, поскольку ряд симптомов актуален не только для этого синдрома, но и многих других болезней. Только после постановки окончательного диагноза можно приступать к терапевтическим мероприятиям.
Лечение синдрома Марфана направлено, в первую очередь, на исправление и профилактику патологий со стороны систем человеческого организма. Особенное внимание уделяют сердечно-сосудистой системе, так как ее поражение влечет за собой серьезную угрозу жизни пациента. лечение может быть консервативным или хирургическим. Решение о выборе оптимального способа принимает лечащий врач, ориентируясь на текущее состояние больного.
Нужно понимать, что этот недуг является генетическим, то есть, его нельзя вылечить.
https://youtu.be/5O5F21XWzWo
Пациенту нужно следить за тем, чтобы свести к минимуму риск поражения тех или иных органов, а также устранить уже существующие проблемы. Благодаря этому, существенно повысится качество его жизни.
Хирургическое лечение проводится преимущественно на сердце и сосудах, а также на глазах. При правильном подходе к борьбе с этим синдромом, продолжительность жизни пациента можно увеличить до 70 лет, что является очень хорошим показателем.
Источник: https://vseolady.ru/sindrom-marfana.html
Источник
Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.

Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
Симптомы синдрома Марфана
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
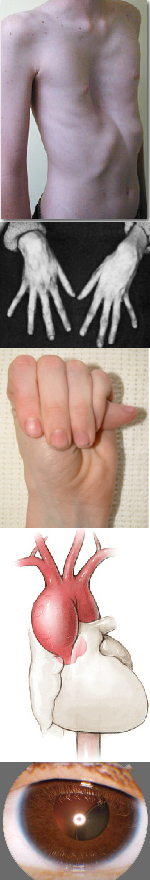 Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз, сколиоз;
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— плоскостопие;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— высокое нёбо;
— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.

Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:

Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн. Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Источники и ссылки:
1. Причины высокого роста — статья, в которой рассмотрены основные причины высокого роста человека.
2. Синдром Марфана в википедии — о синдроме Марфана в свободной энциклопедии.
3. Русскоговорящее сообщество людей с синдромом Марфана — форум о синдроме Марфана, общение, обсуждение болезни.
4. Международный фонд синдрома Марфана — фонд помощи больным с синдромом Марфана.
5. Группа вконтакте для больных синдромом Марфана.
Источник

Синдром Марфана — генетически детерминированное заболевание, характеризующееся поражением или недоразвитием соединительнотканных волокон во время эмбриогенеза и проявляющееся дисфункциональными изменениями со стороны зрительного анализатора, костно-суставной и кардиоваскулярной систем.
Обычно синдром передается по наследству от родителей детям по аутосомно-доминантному принципу. В некоторых случаях он становится результатом мутации гена, кодирующего синтез фибриллина и оказывающего влияние на формирование одновременно нескольких фенотипических признаков. Генетические изменения – мутации происходят под воздействием негативных эндогенных и экзогенных факторов. Белок фибриллин является важной составной частью многих структур организма. При его недостатке соединительная ткань теряет свою прочность и эластичность, что отражается на состоянии сосудистой стенки и связочно-суставного аппарата.
Синдром был открыт в 1875 году офтальмологом из Америки Э. Вильямсом. Он обнаружил полное смещение хрусталика глаза от своего нормального положения у брата и сестры, которые имели высокий рост и чрезмерную подвижность суставов с рождения. Спустя несколько лет детский врач из Франции А. Марфан вел наблюдения за девочкой 5 лет с прогрессирующими аномалиями скелета, чрезмерно длинными конечностями и «паучьими пальцами». Он дал четкое описание патологии, благодаря чему синдром получил его имя.

мальчик с синдромом Марфана
Клиническая симптоматика синдрома весьма полиморфна. Это связано с наличием пораженной соединительной ткани во всех внутренних структурах организма. Системная соединительнотканная недостаточность проявляется признаками поражения скелета, сердца и сосудов, глаз, кожи, ЦНС, легких. Подобные патологические изменения формируются внутриутробно у плода. Симптоматика синдрома варьируется от стертых форм до процессов, несовместимых с жизнью.
Больные имеют диспропорциональные конечности, рост выше среднего, вытянутые пальцы, гипермобильные суставы, худощавое тело, готическое небо, неправильный прикус, глубоко расположенные глаза. Они страдают гигантизмом, миопией, эктопией хрусталика, изменением формы грудины, кифосколиозом. Клинические признаки синдрома обусловлены гиперрастяжимостью тканей.

Синдром Марфана — наследственная коллагенопатия, встречающаяся у 1 из 10000-20000 человек. Патология обнаруживается повсеместно, среди людей любой национальности, пола и места проживания – одинаково часто как среди жителей юга, так и севера. В семейных парах, где отцу больше 35 лет, чаще рождаются больные дети.
Диагностика патологии основывается на данных наследственного анамнеза, визуального осмотра, результатах дополнительных исследований. Пациенты с синдромом Марфана внешне похожи друг на друга. Специалисты по внешнему виду могут предположить наличие недуга. Лечение болезни медикаментозное и оперативное, заключающееся в устранении структурных и функциональных нарушений в сердечной мышце, зрительном анализаторе, костно-суставном аппарате. Оперативное вмешательство показано в тех случаях, когда лекарственное и физиотерапевтическое лечение не дает положительных результатов. Если недуг не лечить, продолжительность жизни больных ограничится 30—40 годами. Причиной их смерти становится разрыв аорты или острая коронарная недостаточность. Современная медицина позволяет пациентам успешно лечиться и полноценно жить до самой старости.
Этиологические факторы
Синдром Марфана – врожденная аномалия, отличающаяся следующими генетическими признаками:
-
 аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;
аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении; - выраженным плейотропизмом — множественным действием гена;
- варьирующей экспрессивностью – степенью развития признака, контролируемого данным геном;
- высокой пенетрантностью – вероятностью фенотипического проявления признака при наличии соответствующего гена.
Синдром развивается в результате мутации гена, кодирующего биосинтез особого белка фибриллина — важной структуры межклеточного вещества, которая обеспечивает эластичность и сократительную способность соединительнотканных волокон. Мутация происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде. При недостатке фибриллина нарушается процесс формирования волокон. Он перестают быть прочными и упругими, становятся чрезмерно растяжимыми и менее устойчивыми к деформациям. В наибольшей степени повреждению подвержены сосуды и связки.
Фибриллин необходим для работы цинновой связки, с помощью которой хрусталик прикрепляется к ресничному телу. При дефиците белка эта связка ослабляется, что проявляется миопией, подвывихом хрусталика, вторичной глаукомой, снижением остроты зрения. Кроме зрительного анализатора, белок фибриллин содержится в связках аорты и обеспечивает ее устойчивость к нагрузкам. При ослаблении этих связок происходит расширение сосуда и расслоение его стенок. Подобные изменения являются смертельно опасными для человека. При синдроме Марфана часто отмечается поражение двустворчатого клапана, что требует проведения хирургической коррекции.
Классификация
Формы патологии:
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
Этиологическая классификация:
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Симптоматика
Синдром Марфана проявляется разнообразными клиническими признаками, что связано с присутствием соединительнотканных волокон в различных структурах организма. У больных появляются признаки поражения костей и суставов, зрительного анализатора, сердца, сосудов, нервов, легких, кожного покрова.
Симптомы дисфункциональных расстройств внутренних органов возникают на фоне сильной усталости и быстрой утомляемости после незначительных физических нагрузок, миалгии, вялости, мышечной гипотонии, приступообразной цефалгии. По мере роста и развития организма человека черты заболевания становятся более выраженными.

Скелет
Клинические признаки синдрома Марфана, обусловленные поражением костей, мышц и связок:
- короткое туловище и длинные ноги,
- молоткообразные пальцы стопы,
- паучьи пальцы кисти,
- худощавое телосложение,
- гипотонус мышц,
- удлиненное и узковатое лицо,
- глубокая посадка глаз,
- отсутствие зазоров между зубами,
- большой нос,
- микрогнатия,
- недоразвитые скулы,
- большие и низко расположенные уши,
- «готическое» небо,
- прогения,
- гиперподвижность суставов,
- деформированная грудная клетка в виде воронки или киля птицы,
- искривление позвоночника,
- смещение вышележащего позвонка относительно нижележащего,
- уплощение сводов стопы,
- продавливание вертлужной впадины.

Сердце и сосуды
Поражение сердечной мышцы при синдроме Марфана определяет его исход. Обычно возникают дефекты в структуре аорты, легочного ствола, развиваются пороки развития клапанов сердца:
- дилатация предсердий и желудочков,
- мешкообразное расширение ограниченного участка аортальной стенки,

- пролабирование двустворчатого клапана,
- миксоматоз сердца,
- кардиомиопатия,
- разрыв хорд митрального клапана,
- кальциноз аортального клапана,
- различные виды аритмии – мерцательная, экстрасистолия,
- воспаление внутренней оболочки сердца – эндокарда,
- кардиалгия с иррадиацией в спину, ключицу, руку, плечо,
- похолодание конечностей,
- затрудненное дыхание,
- сердечные шумы,
- на ЭКГ — признаки ИБС.
Глаза
У лиц с синдромом Марфана развивается патология зрения:
- миопия,
- смещение хрусталика в сторону,
- уплощение роговой оболочки глаза и ее утолщение,
- недоразвитие радужки и ресничной мышцы,
-
 страбизм,
страбизм, - катаракта,
- асимметричность зрачков,
- глаукома,
- астигматизм,
- дальнозоркость,
- колобома глаза,
- спазмирование сосудов сетчатки и высокий риск ее отслойки.
Прочие органы
Поражение нервной системы:
- ишемические и геморрагические инсульты,
- субарахноидальное кровотечение,
- выпячивание оболочек спинного мозга.
Ослабление и растяжение дурального мешка проявляется болью в пояснице, ногах, тазу и другими неврологическими признаками, цефалгией.
Поражение бронхопульмональной системы:
- скопление воздуха между висцеральным и париетальным листками плевры,
- эмфизема легких,
- удлинение и перерастяжение альвеол,
- ночное апноэ,
- рак легких.

У больных пневмотораксом воздух скапливается в плевральной полости, а легкие сжимаются. Их дыхание становится частым и поверхностным, в груди болит, кожные покровы сначала бледнеют, а затем приобретают синюшный оттенок.
Поражение кожи, мягких тканей, внутренних органов:
- смещение почки в каудальном направлении,
- цистоцеле,
- пролапс матки,
- кисты и новообразования в печени и почках,
- варикоз,
- стрии на коже,
- грыжи.
Больные с синдромом Марфана легко возбудимы, гиперактивны, чрезмерно эмоциональны, часто плаксивы и разражительны. Они имеют неординарные способности и высокий уровень интеллекта.
«Готическое» небо и микрогнатия приводят к возникновению речевых нарушений. При поражении скелета и суставов появляются артралгии и миалгии, развивается ранний остеоартрит.
Синдром Марфана проявляется по-разному у лиц, обращающихся за медицинской помощью. У одних выявляются ярко выраженные признаки патологии, у других симптоматика бывает стертой. Прогрессирование синдрома происходит по мере взросления человека. Клинические проявления определяются локализацией и интенсивностью поражения органов и систем.
Методы диагностики

проявления синдрома Марфана в младшем возрасте
Выявлением синдрома Марфана занимаются специалисты в области генетики, кардиологии, офтальмологии, неврологии, ортопедии. Диагностика патологии включает сбор анамнеза жизни и болезни, выявление типичных клинических признаков, анализ внешнего осмотра и физикальных данных, результатов кардиографического и рентгенографического обследования, посещения офтальмолога, составление родословной у генетика.
Основные диагностические методики:
- общий анализ крови и мочи — типичные признаки воспаления;
- биохимическое исследование крови позволяет выявить дисфункцию определенного органа, возникшую в результате развития патологического процесса, а также определить первопричину заболевания и назначить правильное лечение;
- электрокардиография отображает электрические потенциалы, сформированные в работающем сердце;
- эхокардиография – исследование морфологических и функциональных изменений сердца и его клапанного аппарата;
- рентгенографическое и томографическое исследование — информативные диагностические методики, обнаруживающие поражение костей, суставов, внутренних органов и мягких тканей;
- аортография – рентгенологического исследования аорты с использованием контрастного вещества;
- УЗИ внутренних органов,
- биомикроскопия и офтальмоскопия,
- молекулярно-генетический анализ.
Методы лечения

Синдром Марфана неизлечим. Этиотропной терапии недуга не существует, ведь невозможно заменить гены ребенка. Больным проводят симптоматическую терапию, целью которой является облегчение общего состояния, устранение симптомов и предупреждение тяжелых осложнений.
Медикаментозное лечение:
- β-адреноблокаторы – «Пропранолол», «Атенолол»;
- блокаторы кальциевых каналов – «Нифедипин», «Верапамил»;
- ингибиторы АПФ – «Каптоприл», «Лизиноприл»;
-
 коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»;
коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»; - метаболики – «Рибоксин», «Милдронат»;
- поливитаминные комплексы;
- антибиотики для профилактики инфекционного эндокардита;
- антикоагулянты для профилактики тромбоза;
- антиоксиданты и антигипоксанты – «Коэнзим Q10», «Элькар»;
- ноотропные препараты – «Пирацетам», «Винпоцетин».
При снижении остроты зрения пациентам с офтальмологическими заболеваниями прописывают очки для постоянного ношения или контактные линзы.
Хирургическое лечение:
- оперативное вмешательство на сердце и аорте — протезирование аорты и клапанов сердца, реконструктивные и пластические операции,
- операции на глазах — коррекция близорукости лазером, замена хрусталика, устранение глаукомы,
- хирургическая коррекция скелета — пластика грудной клетки при ее воронкообразной деформации, стабилизация позвоночника, протезирование крупных суставов.
Людям с марфаноидным фенотипом стертой формы показано физиолечение и ЛФК.
Клинические рекомендации специалистов своим пациентам:
- ведение здорового образа жизни,
- ограничение тяжелого труда и физического перенапряжения,
- отказ от спортивных игр повышенной активности,
- регулярное посещение специалистов в области кардиологии, офтальмологии, ортопедии, неврологии,
- нахождение больных под постоянным наблюдением и контролем врачей,
- периодическое прохождение диагностического обследования.
Прогноз и профилактические мероприятия
