Синдром прадера вилли у девочек

Синдром Вилли-Прадера – это врожденное генетическое заболевание, которое может встречаться у девочек и мальчиков. Причины связаны с отсутствием или недостаточным функционированием участков генов на 15 отцовской хромосоме. В зависимости от степени «поломки» отличаются клинические проявления. Они могут быть незначительными или с классической картиной ожирения, умственной отсталости и нарушением полового созревания.
Как проявляется патология
До рождения ребенка заподозрить патологию нельзя. Исключение составляют случаи, когда в семье рождались дети с этим отклонением, и доказана его наследственная природа. У остальных поломка в хромосоме является случайной. Причины этого не установлены.
Синдром Вилли-Прадера является врожденной патологией
Внутриутробное развитие ребенка не страдает, но он может родиться с дефицитом веса, гипотрофичным. Во время беременности женщина может отмечать низкую подвижность плода. К родам дети часто занимают неправильное положение, могут находиться в ягодичном предлежании. Иногда рождаются в состоянии гипоксии. Сниженный мышечный тонус после рождения сохраняется в течение первого года жизни. Для них характерны следующие симптомы:
- сниженный сосательный рефлекс;
- задержка формирования двигательных функций;
- поздно начинают держать голову;
- с трудом учатся сидеть, ползать;
- поздно учатся ходить.
Неукротимый голод появляется к 3-4 году жизни. Ребенок много ест и не наедается. Это связано с нарушением выработки гормона грелина, который стимулирует аппетит. У ребенка развивается ожирение, на фоне которого возможны осложнения:
- ночное апноэ (эпизоды остановки дыхания во сне);
- сколиоз;
- артериальная гипертензия.
Задержка умственного развития проявляется в дошкольном возрасте. Дети с синдромом Вилли-Прадера имеют хорошую зрительную память, учатся читать, имеют большой словарный запас, но не используют его в повседневной жизни. Плохо развиты навыки письма, математическая память и восприятие на слух.
В период полового созревания наблюдается отставание от возрастных норм, что связано с гипогонадизмом. Уже в момент рождения у мальчиков отмечается уменьшенный размер пениса, крипторхизм. У девочек часто встречается гипоплазия матки и яичников, аномалии развития наружных половых органов.
В большинстве случаев взрослые с синдромом Вилли-Прадера бесплодны. Но вторичные половые признаки могут появляться при использовании заместительной терапии в период полового созревания.
У синдрома Вилли-Прадера множество разнообразных проявлений, но у одного человека присутствует сочетание только нескольких из них. В тяжелых случаях могут наблюдаться судорожные припадки, признаки нарушения координации движений. Такое многообразие проявлений часто затрудняет постановку диагноза.
Внешний вид ребенка с синдромом Вилли-Прадера
Дети с врожденной генетической патологией отличаются по внешнему виду от нормально развитых, но максимальные различия становятся заметны в подростковом возрасте. Для них характерны:
- узкая височная кость;
- долихоцефалия – удлиненная голова;
- миндалевидный разрез глаз и косоглазие;
- широкая переносица;
- низко посаженные ушные раковины;
- маленький рот и тонкая верхняя губа;
- непропорционально мелкие кисти и стопы;
- низкорослость;
- ожирение.
Иногда у мальчиков или девочек может произойти скачок роста в возрасте 8-9 лет из-за повышения уровня андрогенов, но позже происходит быстрое закрытие зон роста на трубчатых костях, поэтому дети отстают от средних показателей роста для своего возраста. У большинства больных нарушается выработка кожного пигмента, поэтому кожа выглядит бледной, глаза светлые, могут быть слабо пигментированные волосы.
По характеру дети доброжелательны, но страдают от частой смены настроения. Позже могут развиваться обсессивно-компульсивные расстройства, повышенное упрямство.
Отмечается снижение физической активности из-за недостаточного развития мышечной массы и гипотонии.
Дети сонливы, невнимательны, предпочитают сидячие занятия или просмотр телевизора любым другим играм.
Опасные осложнения
Ожирение связано с неконтролируемым аппетитом. Дети с 4 лет едят и не чувствуют насыщения. Это состояние плохо поддается коррекции при помощи диеты, запреты и неудовлетворение потребности ребенка приводят к повышенной невротизации, ухудшению отношений с родителями.
Неконтролируемое потребление пищи может закончиться морбидным ожирением, которое приводит к тяжелым осложнениям. От избыточной массы тела уже в детском возрасте возникает нарушение дыхания, ночью регистрируются эпизоды апноэ. Такие дети склонны к частым респираторным инфекциям. Гиповентиляция легких приводит к тяжелому течению болезней, развитию пневмонии, которая может привести к летальному исходу.
Симптомы: склонность к перееданию, ожирение и задержка развития
Часто развивается сахарный диабет 2 типа, связанный с нарушением чувствительности клеток к инсулину. При несоблюдении диеты на фоне патологии развивается гипергликемия, которая ведет к поражению органов-мишеней:
- головной мозг;
- почки;
- сердце;
- сосуды.
Рано появляются признаки полинейропатии, снижается острота зрения, проявляется нефропатия. Дети с раннего возраста страдают от артериальной гипертензии. Это усугубляет состояние сердца, которое испытывает дополнительную нагрузку. По результатам обследования выявляется гипертрофия (увеличение размеров) левого желудочка.
Методы диагностики
Диагностика основывается на генетическом исследовании, которое позволяет выявить поломку в хромосомах и исключить синдром Дауна, который часто диагностируется ошибочно. Исследование рекомендовано всем новорожденным со сниженным мышечным тонусом, который не устраняется в течение первого месяца жизни.
Остальные методы диагностики направлены на выявление патологических изменений в других органах. Необходимы следующие методы обследования:
- кровь на глюкозу – для раннего выявления признаков диабета, при подозрении на него – дополнительно гликированный гемоглобин и глюкозотолерантный тест;
- определение уровня соматотропина – для выявления причин низкорослости и подбора терапии;
- УЗИ внутренних органов – для раннего выявления последствий ожирения;
- ЭКГ – для контроля работы сердца;
- УЗИ малого таза у девочек – выявление состояния матки и яичников;
- анализ на половые гормоны в подростковом возрасте – с целью коррекции полового созревания и контроля лечения.
Для исключения патологии гипоталамо-гипофизарной области и связанной с этим низкорослостью делается МРТ головного мозга.
Что включает в себя лечение
Болезнь является генетически детерминировнной, поэтому полное излечение невозможно. Но хорошие прогнозы врачи дают тем детям, у которых патология диагностирована на ранних этапах. Новорожденным с гипотонией назначается курс массажа и лечебной физкультуры. Рекомендуется грудное вскармливание, как наиболее сбалансированное питание для младенца. Прикормы вводятся строго по возрасту, начиная с овощных пюре.
Детям старше года необходимо прививать правильное пищевое поведение для профилактики переедания в будущем. Не нужно вводить в рацион сладкое, выпечку, мучное, чтобы они не становились основой меню. Позже рекомендуется питание на основе овощей, фруктов, злаков, с пониженной калорийностью. Ограничивать жирные продукты, не жарить. Диета должна быть строгой, тщательно контролироваться родителями.
В детском возрасте не используются препараты для подавления аппетита, а также нарушающие всасывание липидов и углеводов в кишечнике.
Основу патогенетической терапии составляет назначение гормона роста. Его начинают использовать в минимальной дозировке у детей до года. Это оказывает положительное влияние на моторное развитие, уменьшает признаки гипотонии мышц. Также наблюдаются следующие положительные эффекты от терапии:
- увеличение основного обмена и расхода энергии;
- повышение физической силы и выносливости;
- увеличение мышечной массы;
- улучшение дыхательной функции за счет прироста силы инспираторных (дыхательных) мышц;
- снижение продолжительности и количества эпизодов апноэ.
Многие исследователи отмечают, что на фоне терапии гормоном роста у детей улучшается поведение. Они становятся более внимательными и активными. Но лечение прекращают при отсутствии эффекта или при прогрессировании ожирения. Гормон роста способствует повышению уровня инсулина и инсулинорезистентности, поэтому не рекомендуется детям с нарушением толерантности к глюкозе. Другие методы терапии направлены на коррекцию сопутствующих заболеваний.
Профилактика врожденного синдрома невозможна. Но чтобы предотвратить повторное рождение детей с такой аномалией, семейным парам рекомендуется медико-генетическое консультирование и обследование. Синдром Вилли-Прадера может быть как спонтанной мутацией, так и наследуемой патологией.
Смотрите далее: синдром вильямса
Источник
Синдром Пра́дера — Ви́лли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг. Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование. Частота встречаемости — 1 : 12 000-15 000 живорождённых младенцев. Патология встречается с одинаковой частотой у женщин и мужчин[2].
Наиболее частой причиной синдрома (70-75 % случаев) является делеция участка 15q11-13 хромосомы 15, унаследованной от отца. Около четверти случаев обусловлено однородительской дисомией хромосомы 15 upd(15)mat, когда обе 15-е хромосомы у пациента являются копиями материнского происхождения. В незначительном числе случаев синдром связан с нарушением импринтинга или с наличием сбалансированной транслокации с точкой разрыва внутри участка 15q11-13[3].
Синдром впервые описали в 1956 году учёные из Швейцарии А. Прадер (A. Prader), Х. Вилли (H. Willi) и А. Лабхарт (A. Labhart)[4].
Особенности[править | править код]
Для синдрома Прадера — Вилли характерны:
- до рождения — низкая подвижность плода, часто — неправильное положение плода;
- дисплазия тазобедренных суставов;
- ожирение; склонность к перееданию (чаще проявляется к двум годам);
- пониженный мышечный тонус (гипотонус); пониженная координация движений;
- маленькие кисти и стопы, низкий рост;
- повышенная сонливость;
- страбизм (косоглазие);
- сколиоз (искривление позвоночника);
- пониженная плотность костей;
- густая слюна; плохие зубы;
- сниженная функция половых желёз (гипогонадизм) и в результате, как правило, бесплодие;
- речевая задержка, задержка психического развития; отставание в освоении навыков общей и мелкой моторики.
- более позднее половое созревание.
Внешние признаки: у взрослых выражена переносица; лоб высокий и узкий; глаза, как правило, миндалевидные; губы узкие.
Как правило, у больного встречается не более пяти вышеуказанных признаков.
Диагностика[править | править код]
Синдром диагностируется путём генетического анализа, рекомендуемого для новорождённых с пониженным мышечным тонусом (гипотонусом).
Иногда вместо диагноза «синдром Прадера — Вилли» врачи ошибочно ставят диагноз «синдром Дауна» (поскольку синдром Дауна встречается намного чаще).
Дети с синдромом Прадера — Вилли очень похожи между собой, опытный генетик сможет быть уверен в диагнозе, не дожидаясь результатов исследования кариотипа.
Лечение[править | править код]
Синдром Прадера — Вилли является врождённой генетической аномалией; в настоящее время специфические способы его лечения не разработаны.
Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии. Рекомендуются использование специальных методик развития ребёнка, занятия с логопедом и дефектологом. Показан приём «гормонов роста», заместительная гормональная терапия (с применением гонадотропинов).
Гипогонадизм обычно проявляется в микропении (микропенис — половой член аномально малого размера) и неопущении яичек у мальчиков (крипторхизм); врачи могут посоветовать подождать, пока яички опустятся сами, или порекомендовать хирургическое вмешательство либо гормонотерапию.
Для коррекции избыточной массы тела применяется диета с ограничением количества жиров и углеводов. Из-за ожирения, сопутствующего синдрому, нужно пристально следить за количеством и качеством пищи, поглощаемой человеком с синдромом Прадера — Вилли (обычно люди с таким синдромом способны много съесть, не наедаясь).
Возможным осложнением может стать апноэ (задержка дыхания во сне).
Риск[править | править код]
Риск, что следующий ребёнок у тех же родителей родится также с синдромом Прадера — Вилли, зависит от механизма, вызвавшего генетический сбой.
Этот риск меньше 1 %, если у первого ребёнка делеция гена или партеногенетическая (однородительская) дисомия; составляет до 50 %, если сбой вызван мутацией; до 25 % — в случае транслокации родительских хромосом. Родителям рекомендуется пройти генетическое обследование.
Перспективы развития[править | править код]
У большинства людей с синдромом Прадера — Вилли наблюдается задержка психического и речевого развития.
Согласно исследованиям, которые провели Л. М. Керфс и Дж. П. Фринс (1992)[5],
- 5 % обследованных продемонстрировали средний уровень коэффициента интеллекта (более 85 баллов по шкале IQ);
- 27 % — уровень на грани среднего (70-85 баллов);
- 34 % — уровень слабого отставания (50-70 баллов);
- 27 % — уровень среднего отставания (35-50 баллов);
- 5 % — сильное отставание (20-35 баллов);
- менее 1 % — значительное отставание.
По другим исследованиям (Кэссиди), 40 % пациентов с синдромом Прадера — Вилли демонстрируют интеллект на грани среднего или сниженный уровень интеллекта.
Как правило, дети с синдромом Прадера — Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарём, но их собственная речь обычно хуже, чем понимание.
Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже.
Синдром Прадера — Вилли нередко ассоциируется с повышенным аппетитом. У больных повышена концентрация в крови гормона грелина. Для них также характерна пониженная концентрация соматолиберина. Это обусловлено тем, что 15-я хромосома связана с гипоталамусом. Однако при вскрытии умерших с синдромом Прадера — Вилли не было обнаружено никаких дефектов гипоталамуса. По другим данным, наблюдалось снижение общего количества клеток и окситоцин-содержащих клеток паравентрикулярных ядер гипоталамуса[6]
См. также[править | править код]
- Синдром Мёбиуса
- Синдром Ангельмана
Примечания[править | править код]
Ссылки[править | править код]
- Л. З. Казанцева, П. В. Новиков, А. Н. Семячкина, Е. А. Николаева, М. Б. Курбатов, Э. В. Добрынина. Синдром Прадера — Вилли у детей: новое в этиологии, патогенезе и лечении. Московский НИИ педиатрии и детской хирургии Минздрава РФ / Рос. вестник перинатологии и педиатрии. — 1999. — № 4. — С.40-44.
- Сайт «Биология человека»
- Однородительская дисомия на сайте «Биология человека»
- О синдроме на сайте «Эндокринология» (EURODOCTOR.ru — Элитное лечение в Европе)[неавторитетный источник?]
- Форум родителей детей с этим диагнозом
- Мэтт Ридли. Геном: автобиография вида: В 23 главах. Глава из книги (Matt Ridley. Genome: The Autobiography of a Species in 23 Chapters)
- Подборка статей по теме, общение родителей (форум)
- Польза и вред от гормонов роста (реферат)[неавторитетный источник?]
- Кармен Сапиенца. Геномный импринтинг // Scientific American. Издание на русском языке. — 1990. — № 12. — С.14-20.
Источник
Что такое синдром Прадера — Вилли?
Синдром Прадера — Вилли (СПВ) — это генетическое заболевание, которое встречается примерно у одного из каждых 15 000 новорожденных.
Синдром Прадера — Вилли поражает мужчин и женщин с одинаковой частотой и затрагивает все расы и этнические группы. СПВ признана наиболее распространенной генетической причиной опасного для жизни детского ожирения.
Данный синдром был впервые описан швейцарскими врачами Андреа Прадером, Алексисом Лабхартом и Генрихом Вилли в 1956 году на основании клинических характеристик девяти обследованных детей.
Общие характеристики, определенные в первоначальном отчете, включали маленькие руки и ноги, аномальный рост и состав тела (маленький рост, очень низкая мышечная масса тела и раннее детское ожирение), мышечная гипотония (низкий мышечный тонус (слабость мышц)) при рождении, ненасытный голод, сильное ожирение и умственная отсталость.
Это видео даст краткое понимание проявлений болезни:
Синдром Прадера — Вилли является результатом аномалии хромосомы 15, и постановка точного диагноза сегодня основывается на генетических анализах.
Каковы симптомы синдрома Прадера — Вилли?
Симптомы синдрома Прадера — Вилли, вероятно, связаны с дисфункцией части мозга, называемой гипоталамус. Гипоталамус — это небольшой эндокринный орган у основания мозга, который играет важную роль во многих функциях организма, включая регулирование голода и сытости, температуры тела, боли, баланса сна и бодрствования, водного баланса, эмоций и фертильности.
Хотя считается, что дисфункция гипоталамуса приводит к появлению симптомов синдрома Прадера — Вилли, еще не ясно, как генетическая аномалия вызывает дисфункцию гипоталамуса.
У людей с данным синдромом симптомы со временем меняются. В целом, есть два основных этапа симптомов, связанных с СПВ:
Ранний период жизни
Младенцы с СПВ являются гипотоническими или «гибкими», с очень низким мышечным тонусом. Для них типичен слабый крик и плохой сосательный рефлекс. Детей с синдромом Прадера — Вилли обычно не могут кормить грудью и они часто нуждаются в искусственном вскармливании.
У детей могут быть проблемы с ростом и развитием, если трудности с кормлением не подвергаются тщательному мониторингу и лечению. По мере взросления этих детей сила и мышечный тонус в целом улучшаются. Моторные функции развиты, но, как правило, выполняются с задержкой.
Малыши, как правило, вступают в период, когда они могут легко начать набирать вес до того, как проявят повышенный интерес к еде.
Детство и взросление
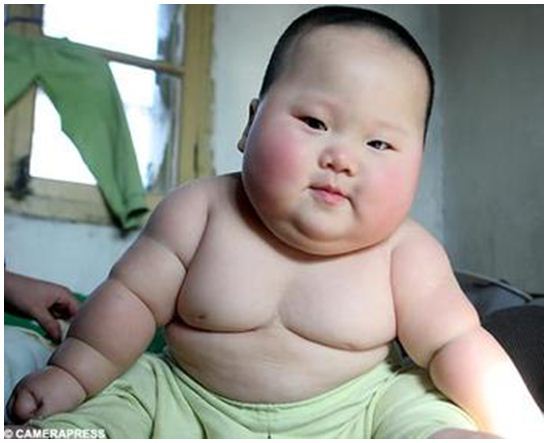
Нерегулируемый аппетит и легкое увеличение веса (см. фото) характеризуют более поздние стадии синдрома Прадера — Вилли.
Эти признаки чаще всего начинаются в возрасте от 3 до 8 лет, но различаются по началу и интенсивности.
Люди с синдромом Прадера — Виллии испытывают недостаток в нормальных подсказках голода и сытости.
Как правило, они не могут контролировать потребление пищи и переедают, если их не контролировать.
Поведение в голоде очень распространено. Кроме того, скорость метаболизма у людей с данным синдромом ниже, чем обычно. Без лечения эта комбинация проблем приводит к патологическому ожирению и его многочисленным осложнениям.
Помимо ожирения, у ребенка с синдромом Прадера — Вилли может быть множество других симптомов. Дети с IQ от низкого нормального до умеренной умственной отсталости обычно имеют когнитивные проблемы. Те, у кого нормальный IQ, обычно имеют проблемы с обучаемостью.
Другие проблемы и признаки могут включать:
- дефицит гормона роста/низкий рост;
- маленькие руки и ноги;
- сколиоз;
- нарушения сна с чрезмерной дневной сонливостью;
- высокий болевой порог;
- апраксия/диспраксия речи и бесплодие.
Поведенческие трудности могут включать в себя обсессивно-компульсивные симптомы, проблемы с кожей и трудности с контролем эмоций. Взрослые с синдромом Прадера— Вилли подвергаются повышенному риску психических заболеваний. СПВ является расстройством широкого спектра, и симптомы варьируют по степени тяжести и частоты встречаемости среди людей.
Хромосомы и гены: основы
Чтобы понять генетику СПВ, необходимо иметь общее представление о хромосомах и генах.
Хромосомы представляют собой крошечные структуры, которые присутствуют почти в каждой клетке нашего тела. Это пакеты генов, которые мы наследуем от наших родителей. Гены содержат все подробные инструкции, необходимые нашему телу для роста, развития и функционирования — нашу ДНК.
Определенные гены направляют наши клетки на производство белков, ферментов и других необходимых веществ. Каждый из наших многочисленных генов находится на определенной хромосоме. Большинство клеток нашего тела содержат 46 хромосом — 23 унаследованы от нашей матери и 23 от нашего отца. (Яйца и сперматозоиды обычно содержат всего 23 хромосомы, потому что это те клетки объединяются в зачатии и обеспечивают ребенку нужное количество хромосом.)
22 пары хромосом помечены числом, основанным на их размере (хромосома 1 — самая большая пара, а хромосома 22 — почти наименьшая), и 2 хромосомы в каждой пронумерованной паре содержат одинаковые гены (один набор у матери и одну от отца). Изменения, которые вызывают синдром Прадера — Вилли, происходят в паре, известной как хромосома 15. 23-я пара хромосом обозначена как пара половых хромосом. Эта пара определяет пол ребенка: XX для девочки, XY для мальчика.
Изменения или ошибки в генах и хромосомах распространены при образовании яйцеклеток и сперматозоидов. Некоторые из этих генетических изменений не будут иметь эффекта, когда ребенок зачат; некоторые вызовут выкидыш; а некоторые спровоцируют, например, синдром Прадера — Вилли, вызвав значительные различия в том, как ребенок развивается и функционирует.
В то время как многие генетические нарушения вызваны изменением одного гена и могут передаваться от родителя к ребенку, СПВ куда более сложен.
Что вызывает синдром Прадера — Вилли (причины)?
Синдром Прадера — Вилли возникает в результате отсутствия активного генетического материала в определенной области хромосомы 15 (15q11-q13). Обычно люди наследуют одну копию хромосомы 15 от своей матери и одну от своего отца. Гены вызывающие СПВ активны только в хромосоме, полученной от отца.
При синдроме Прадера — Вилли генетический дефект, вызывающий неактивность хромосомы 15 от отца (отцовская хромосома 15), может возникнуть по трем следующим причинам:
- СПВ по исключению: чаще всего часть хромосомы 15, которая была унаследована от отца, отсутствует или удалена в этой критической области. Эта небольшая делеция встречается примерно в 70% случаев и обычно не выявляется при обычном генетическом анализе, таком как амниоцентез.
- СПВ по однородительской дисомии: еще около 30% случаев происходят, когда человек наследует две хромосомы 15 от своей матери и ни один от своего отца. Это тип наследование называется однородительской дисомией (UPD).
- СПВ по импрессии мутации: наконец, в очень небольшом проценте случаев (1-3%) небольшая генетическая мутация при синдроме Прадера — Вилли приводит к тому, что генетический материал отцовской хромосомы 15 хоть и присутствует, но неактивен.
Как эти генетические дефекты вызывают симптомы, наблюдаемые при синдроме Прадера — Вилли?
Хромосомы 15 одни из самых сложных областей генома человека. Хотя были достигнуты значительные успехи в понимании и характеристике генетических изменений, связанных с синдромом Прадера — Вилли, точный механизм, с помощью которого отсутствие функционального генетического материала приводит к симптомам, связанным с СПВ, не понятны.
Ученые активно изучают нормальную роль генетических последовательностей в области СПВ и то, как их потеря влияет на гипоталамус и другие системы организма.
Существуют ли различия в степени тяжести СПВ в зависимости от генетического подтипа?
Могут быть некоторые тонкие различия в признаках СПВ на основе генетического подтипа: например, лица с делециями могут быть светлокожими со светлыми волосами по сравнению с другими членами семьи и быть более восприимчивыми к припадкам и судорогам; а те, у кого есть проблемы с СПВ по однородительской дисомии, могут быть подвержены более высокому риску психических заболеваний в молодом подростковом возрасте.
В целом, однако, существует значительное совпадение между различными генетическими подтипами. Вероятно, что тысячи генов за пределами области СПВ, которые демонстрируют нормальные различия между индивидуумами, также вносят значительный вклад в вариабельность симптомов синдрома Прадера — Вилли между теми, у кого есть расстройство.
Как диагностируется СПВ?
Данный синдром диагностируется с помощью анализа крови, который выявляет генетические аномалии, специфичные для синдрома Прадера — Вилли — так называемый «анализ метилирования ДНК». Метод FISH (флуоресцентная гибридизация in-situ) идентифицирует синдром путем делеции, но не диагностирует другие формы СПВ.
Анализ на метилирование ДНК идентифицирует все типы этого синдрома и является предпочтительным методом исследования для диагностики. Если анализ на метилирование проводится первым, может потребоваться дополнительное исследование, чтобы определить, вызван ли СПВ отцовской делецией, UPD или импринтинговой мутацией.
В случаях, когда существует подозрение на наличие импринтинговой мутации, кровь также может быть взята у родителей.
Каковы риски синдрома Прадера — Вилли в будущем?
СПВ по исключению и UPD являются случайными явлениями и, как правило, не связаны с повышенным риском рецидива в будущих беременностях. В случае импринтинг-мутации синдром Прадера — Вилли может в семье повториться. Семьи с опасениями по поводу их риска СПВ должны поговорить с генетическим консультантом.
Есть ли лекарство от синдрома Прадера — Вилли?
В настоящее время нет никакого лечения от синдрома Прадера — Вилли, и большинство исследований на сегодняшний день было направлено на лечение определенных симптомов данного синдрома.
Для многих людей, затронутых этим расстройством, устранение некоторых из самых сложных аспектов синдрома, таких как ненасытный аппетит и ожирение, будет представлять собой значительное улучшение качества жизни и способности жить независимо.
Источник