Синдром пирсона что это такое

Суть болезни
Синдром Пирсона – тяжелое врожденное заболевание, при котором наблюдаются серьезные нарушения кроветворения (прежде всего выработки эритроцитов) и функции поджелудочной железы. Кроме того, нередко у больных встречаются печеночная и почечная недостаточность, а также нарушения работы желез внутренней секреции.
Синдром Пирсона относится к группе митохондриальных заболеваний. Это означает, что его появление связано с «поломкой» в ДНК – но не в тех молекулах ДНК, которые содержатся в хромосомах в ядре клетки и являются основными носителями наследственной информации, а в тех небольших молекулах ДНК, которые содержатся в митохондриях – особых структурах, являющихся «энергетическими фабриками» клеток.
Частота встречаемости
Синдром Пирсона – очень редкое заболевание. В России диагностированы лишь единичные случаи. В мире на данный момент описано менее 100 случаев синдрома Пирсона.
Синдром Пирсона относится к генетически обусловленным болезням. Однако наследование митохондриальных заболеваний отличается от наследования заболеваний, вызванных «обычными» хромосомными дефектами. Митохондриальная ДНК передается детям только от матери; при этом для проявления болезни необходимо, чтобы число доставшихся ребенку дефектных молекул митохондриальной ДНК было достаточно велико – а это число зависит от множества случайных факторов. Поэтому при синдроме Пирсона генетическое консультирование затруднено. В одной и той же семье могут родиться и клинически здоровые, и больные дети.
Признаки и симптомы
Многие проявления синдрома Пирсона связаны с неспособностью костного мозга больных производить нормальные клетки крови. Так, характерной чертой болезни является сидеробластная анемия. Это значит, что в предшественниках эритроцитов нарушено использование железа для синтеза гемоглобина, и возникают сидеробласты – клетки, где под микроскопом видны гранулы «неиспользованного» железа, расположенные обычно в виде кольца вокруг ядра клетки.
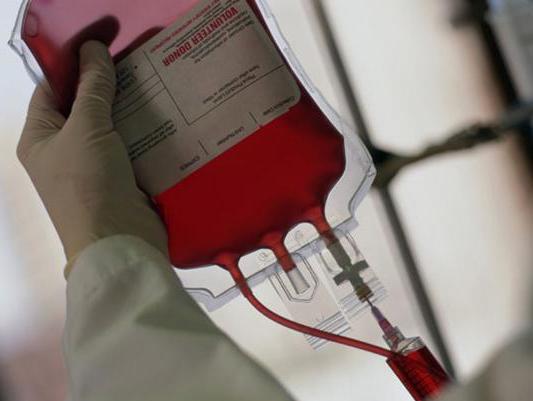
Анемия у таких больных не поддается терапии препаратами железа и витаминами, поэтому бывают необходимы переливания донорских эритроцитов. Нередко наблюдается также низкий уровень тромбоцитов (проявляющийся повышенной склонностью к кровотечениям и образованию синяков) и лейкоцитов (низкая сопротивляемость инфекциям).
Вторая важная черта синдрома Пирсона – недостаточная выработка ферментов поджелудочной железы. В результате пища плохо усваивается, возникает хронический понос. Довольно часто наблюдаются также печеночная недостаточность, проблемы с почками, иногда – нарушения выработки гормонов.
Первые проявления болезни возникают в раннем возрасте, иногда даже с самого рождения. Дети медленно растут и недостаточно набирают вес, плохо развиваются, страдают от постоянного поноса, у них увеличена печень. Время от времени у них случаются метаболические кризы с эпизодами рвоты, сонливости и т.п. Периодически наблюдаются также тяжелые симптомы, связанные с ацидозом, то есть слишком высокой кислотностью крови; при этом страдают многие системы организма, включая желудочно-кишечный тракт, центральную нервную систему, сердце, мышцы, органы дыхания.
Диагностика
При диагностике синдрома Пирсона принимают во внимание как клинические проявления, так и результаты лабораторных исследований. При исследованиях обнаруживается анемия с присутствием значительного количества кольцевых сидеробластов и определенные изменения в клетках костного мозга. Для изучения состояния поджелудочной железы могут использоваться измерения концентрации ее ферментов в сыворотке крови и другие анализы. Измеряется уровень молочной кислоты в сыворотке крови, так как при синдроме Пирсона ее обмен нарушен. Применяются и другие методы исследования.
Разумеется, прямую информацию о наличии синдрома Пирсона можно было бы получить путем генетического анализа митохондриальной ДНК. К сожалению, не всегда этот анализ дает надежные результаты, так как митохондриальная ДНК существует в различных вариантах (это называется гетероплазмией) не только в пределах организма, но даже внутри одной клетки.
Лечение
Специфического лечения синдрома Пирсона не существует. Однако в ряде случаев можно облегчить состояние пациентов и продлить им жизнь.
Из-за анемии больным нужны переливания компонентов крови. Недостаточность поджелудочной железы требует приема панкреатических ферментов. Поскольку у больных нарушен обмен веществ, им бывает необходима инфузионная терапия для коррекции водно-электролитного баланса. Кроме того, так как при синдроме Пирсона обычно наблюдается повышенная кислотность крови, больные получают терапию бикарбонатом натрия или дихлорацетатом. Для купирования инфекционных осложнений используются антибиотики.
Трансплантация костного мозга теоретически может привести к нормализации показателей крови. Но, к сожалению, она не снимает прочие проблемы, возникающие при синдроме Пирсона. Кроме того, при нарушениях, развившихся в результате синдрома Пирсона, сама по себе процедура трансплантации сопряжена с повышенным риском и почти никогда не применяется.
Прогноз
Большинство больных синдромом Пирсона умирает в первые 2-3 года жизни, несмотря на поддерживающее лечение. Однако немногочисленные пациенты проживают более долгую жизнь, причем иногда анемия прекращается сама собой. У таких больных могут развиться признаки синдрома Кернса-Сейра – особой разновидности митохондриальных болезней, которая характеризуется мышечными и другими нарушениями.
Источник
Синдром Пирсона Это одна из известных как редкие болезни, из-за своей низкой распространенности. Он состоит из митохондриального заболевания, которое поражает весь организм, то есть его воздействие является мультисистемным. Его начало происходит в детстве и происходит из-за делеции митохондриальной ДНК.
Этот синдром был впервые описан в 1979 году Говардом Пирсоном, педиатром, специализирующимся на гематологии. Десять лет спустя были обнаружены делеции митохондриальной ДНК, которые вызывают этот синдром..
Причины синдрома Пирсона
Это мультисистемное заболевание вызвано нарушением окислительного фосфорилирования, которое является метаболическим процессом, посредством которого энергия, выделяемая при окислении питательных веществ, используется для производства аденозинтрифосфата (АТФ). Нарушение этого процесса связано с дублированием митохондриальной ДНК.
Несмотря на то, что это болезнь митохондрий, то есть, передаваемая матерью, был сделан вывод о том, что синдром Пирсона обычно является спорадическим. Следовательно, существуют делеции митохондриальной ДНК, которые служат диагностическими критериями, но случайное распределение этого типа ДНК вызывает сближение нормальных клеток, а других с мутациями..
Этот факт, называемый гетероплазмой, который возникает, когда человек представляет смесь разных популяций митохондрий, является причиной большой изменчивости в клинической экспрессии заболевания. Этот термин относится к тому факту, что, несмотря на ответ на один и тот же диагноз, у разных людей проявляются разные симптомы, а также разные уровни аффектации..
Какова его распространенность?
Будучи редким заболеванием, оно поражает меньшинство населения. По данным Европейского портала редких заболеваний, Orphanet, синдром Пирсона имеет распространенность
Кроме того, он добавляет, что описано не более 60 случаев. Тип наследования, передаваемый синдромом Пирсона, поскольку он не связан с полом, одинаково влияет на мальчиков и девочек..
Каковы ваши симптомы?
Синдром Пирсона начинается в младенческой стадии, и есть несколько случаев, описанных у новорожденных. Первые признаки видны в период грудного вскармливания и до шести месяцев жизни.
Этот синдром представляет собой очень разнообразную картину с различными условиями. Есть три характеристики, которые представлены всеми, кто страдает от синдрома Пирсона и которые следующие:
Рефрактерная сидеробластная анемия
Это наиболее существенный симптом синдрома Пирсона, который включает изменение синтеза гемоглобина в предшественниках костного мозга. Таким образом, получают так называемые кольцевые сидеробласты..
Для его лечения удобно контролировать анемию и, кроме того, предотвращать перегрузку железом.
Иногда эта анемия связана с глубокой нейтропенией, которая состоит в уменьшении количества нейтрофилов (обычно известных как лейкоциты или лейкоциты)..
Также от тромбоцитопении; когда есть ненормальная гематологическая ситуация и количество тромбоцитов меньше. Это происходит из-за разрушения ткани эритроцитов в костном мозге.
Вакуолизация предшественников костного мозга
Клетки, которые являются предшественниками костного мозга, в случае синдрома Пирсона, значительно увеличивают свои размеры.
Экзокринная дисфункция поджелудочной железы
Эта дисфункция — неспособность экзокринной поджелудочной железы нормально выполнять пищеварительные функции. Обычно это вызвано внезапным снижением секреции поджелудочной железы. Это тесно связано с плохим пищеварением и, как следствие, приводит к нарушению всасывания непереваренных продуктов, которые часто вызывают состояние недоедания.
Существует выраженная вариабельность в выражении синдрома Пирсона, потому что патогенные клетки находятся вместе с нормальными. Чтобы человек мог представить патологические проявления, он должен накопить достаточное количество мутированной ДНК. Иногда, из-за различных органов и систем, которые поражены, считается, что синдром Пирсона состоит из «бессвязной» ассоциации симптомов.
В публикации Университетской клиники им. Доус де Октубре в Мадриде, в которой было изучено три случая синдрома Пирсона, они показывают, что другие симптомы, которые обычно присутствуют позже, это глазные, эндокринные, сердечные и неврологические заболевания. Что касается сердечных заболеваний, некоторым пациентам требовалась имплантация кардиостимулятора.
В меньшей степени, есть пациенты с диагнозом синдрома Пирсона, у которых есть изменения мозга и / или ствола мозга, которые видны через магнитно-резонансную томографию..
Кроме того, у некоторых из них наблюдается гиперактаторракия, также известная как гипоглюкорраквия, которая предполагает снижение уровня глюкозы в спинномозговой жидкости. Кроме того, гиперпротеинорракия, увеличение белков в спинномозговой жидкости и снижение фолиевой кислоты в этой жидкости являются общими.
Как можно диагностировать синдром Пирсона?
Обычно диагноз может быть поставлен на основании наблюдаемых симптомов. Однако, как указывает Ассоциация синдрома Пирсона, необходимо выполнить различные тесты и экзамены, чтобы сделать вывод о диагнозе этого синдрома..
Во-первых, если есть подозрение на митохондриальный синдром, можно провести профилактический анализ, чтобы определить наиболее частые генетические изменения в митохондриальной ДНК..
Другим очень важным тестом при синдроме Пирсона является биопсия мышц, и в случае, когда различные симптомы сходятся, это очень важно. Этот тест включает в себя удаление небольшого образца мышечной ткани для изучения и анализа. Это быстрый и минимально инвазивный тест, и он также не болезненный.
Нейрорадиология полезна в диагностике этого синдрома, поскольку она предлагает изображения состояния головного мозга, и можно будет обнаружить наличие аномалии. Благодаря лабораторным исследованиям будут измерены уровни молочной кислоты и спинномозговой жидкости, и, таким образом, будет возможно установить, реагируют ли они на средние уровни или есть ли какие-либо отклонения от нормы..
Наконец, но не в последнюю очередь, тесты, которые анализируют активность ферментов.
В тех случаях, когда имеются сердечные симптомы или которые влияют на другие органы или системы, такие как зрение, будут проводиться соответствующие тесты для применения необходимого им лечения. Гастроэнтерологические и диетологические исследования могут также быть выполнены, чтобы проверить, что поглощение питательных веществ выполняется правильно.
лечение
На сегодняшний день синдром Пирсона требует симптоматического лечения. То есть не существует терапии или лекарств для лечения заболевания, и, следовательно, лечение направлено на облегчение симптомов, которые этот синдром вызывает у людей, которые страдают от него..
Для этого, и в первую очередь, очень важно провести исчерпывающий анализ, который позволяет получить данные о состоянии здоровья несовершеннолетнего и его недостатках, чтобы иметь возможность подходить к лечению наиболее подходящим образом. Кроме того, медицинские проверки необходимы, чтобы проверить развитие и убедиться, что используемое лечение является адекватным.
Как правило, лечение будет направлено на облегчение инфекционных эпизодов и метаболических проблем..
В случаях, когда анемия является серьезной, будет предписано переливание крови. В некоторых случаях это лечение будет сопровождаться терапией эритропоэтином, которая заключается в применении гормона, который будет способствовать образованию эритроцитов, также известных как эритроциты..
Также, если таковые имеются, будут лечиться эндокринные нарушения или симптомы, которые затрагивают другие органы, которые не были упомянуты в этом разделе и которые я упоминал выше, такие как зрительная система, сердце и т. Д..
Это смертельно?
К сожалению, синдром Пирсона обычно заканчивает жизнь этих детей до трехлетнего возраста. Причины различны и среди них:
- Риск сепсиса, который является массивной реакцией организма на инфекционный процесс.
- Метаболические кризы с лактоацидозом или гепатоцеллюлярной недостаточностью.
Нет никаких данных, которые бы говорили нам о выживаемости детей, страдающих этим синдромом. Но в случае, если эти несовершеннолетние выживают после симптоматики, синдром Пирсона исчезает из-за фенотипической эволюции, спонтанно исчезая гематологические симптомы.
Что касается неврологических и миопатических признаков, они могут увеличиваться или исчезать. В некоторых случаях синдром Пирсона приводит к другому митохондриальному заболеванию, которое является синдромом Кернса-Сэйра.
Что такое синдром Кернса-Сайре??
Этот синдром, также митохондриального типа, характеризуется прогрессирующей наружной офтальмоплегией (прогрессирующая слабость глазных мышц и подъемов век), пигментным ретинитом (группировкой дегенеративных заболеваний глаз) и его начало наступает до 20 лет. Некоторые дополнительные общие черты включают глухоту, мозжечковую атаксию и сердечную блокаду.
Цифры его распространенности, предложенные Orphanet, показывают, что синдром Кернса-Сэйра поражает одного человека из 125 000.
Обычно заболевание проявляется в младенческой стадии со следующими симптомами: птоз (полное или частичное отделение органа), пигментная ретинопатия и прогрессирующая внешняя офтальмоплегия. Затем появляются другие симптомы в зависимости от распределения молекулярной аномалии, как при синдроме Пирсона.
Другими симптомами, связанными с этим синдромом, являются двусторонняя нейросенсорная глухота, сердечные поражения, поражения центральной нервной системы (мозжечковая атаксия, дизартрия, двусторонняя слабость лица, умственный дефицит), миопатия скелетных мышц, кишечные и эндокринные расстройства (замедленное половое созревание) , гипопаратиреоз, диабет) и почечная недостаточность. Прогрессирование заболевания происходит медленно и может длиться до десятилетий. В эти годы могут появиться новые симптомы или ухудшиться те, которые уже присутствуют.
Синдром Кернса-Сайре также вызван делециями фрагментов митохондриальной ДНК, влияющими на процесс окислительного фосфорилирования. Есть исключительные случаи этого синдрома, которые происходят без удаления митохондриальной ДНК и являются следствием точечных мутаций, расположенных в той же самой.
Диагноз обычно ставится на основании проявлений и, впоследствии, испытаний, проводимых для его подтверждения. Тесты обычно такие же, как и в случае синдрома Пирсона. Обычно диагноз не ставится на дородовой стадии.
Большинство случаев этого синдрома происходят спорадически. Исключения митохондриальной ДНК передаются исключительно от одного поколения к другому. По оценкам, менее 4% женщин передают свое потомство на делецию митохондриальной ДНК. В случае мужчин, они не передают это.
Таким же образом, лечение этого синдрома пытается облегчить спровоцированные симптомы. Рекомендуются регулярные осмотры у кардиолога. В случаях, когда возникают сердечные блоки, им потребуется имплантация кардиостимулятора или устройства, которое дефибриллятор, чтобы улучшить качество жизни этих пациентов.
Глухие пациенты могут использовать слуховые аппараты. Кроме того, было обнаружено, что добавка коэнзима Q10 является полезной в некоторых случаях. В случае офтальмологических проявлений их можно лечить хирургическим путем, хотя риск рецидива, а также риск возможных глазных осложнений высок..
Прогноз человека, страдающего синдромом Кернса-Сайре, будет зависеть от пораженных органов и степени вовлеченности в каждый из них. Этот факт тесно связан с долей митохондриальной ДНК, пораженной и здоровой, которая находится в каждой из них..
В большинстве случаев ожидаемая продолжительность жизни людей, страдающих этим синдромом, может быть нормальной, если они получают адекватную медицинскую помощь в соответствии с лечением и рекомендациями, предписанными медицинскими работниками..
библиография
- McShane, M.A. (1991) Синдром Пирсона и митохондриальная энцефаломиопатия у пациента с делецией мтДНК. Отделение неврологии, больница для больных детей, Queen Square, Лондон.
- Синдром Кернса-Сайре. Сирота (2014).
- Синдром Пирсона. Сирота (2006).
- Cánovas, R. de la Prieta, J.J. Алонсо, К. Руис, Т. Перейра, К. Агирре. Сидеробластные анемии (2001). Сервис и кафедра внутренних болезней. УПВ / ЕГУ. Больница Cruces. Barakaldo.
- Мартин Эрнандес, М.Т. Гарсия Силва, П. Кихада Фрайле, А. Мартинес де Арагон, А. Кабелло, М.А. Мартин. Синдромы Пирсона и Кернса-Сайре: две мультисистемные митохондриальные болезни, обусловленные делециями в митохондриальной ДНК (2010).
- Каммарата-Скалиси, Ф., Лопес-Галлардо, Е., Император, С., Руис-Песини, Е., Да Сильва, Г., Камачо, Н., Монтойя, Дж.. Синдром Пирсона. Отчет по делу (2011).
Источник
Синдром Пирсона – это очень редкое генетическое заболевание, которое проявляется еще в младенчестве и в большинстве случаев ведет к раннему летальному исходу.
История открытия
Другое название синдрома Пирсона – врожденная сидеробластная анемия с экзокринной недостаточностью поджелудочной железы. Болезнь названа в честь ученого, впервые ее описавшего в 1979 году – Н. А. Пирсона. Синдром был распознан благодаря длительным наблюдениям за четырьмя детьми со схожими симптомами: у них наблюдалась сидеробластная анемия, которая не поддавалась стандартному лечению, недостаточность экзокринной функции поджелудочной железы и патология клеток костного мозга.
Сначала детям ставили другой диагноз – синдром Швахмана (врожденная гипоплазия поджелудочной железы). Но после исследования крови и костного мозга были выявлены явные отличия, что и дало повод выделить Пирсона синдром в отдельную категорию.
Причины возникновения болезни
Исследование причин заболевания заняло около десяти лет. Врачи-генетики сумели найти генетический дефект, который ведет к делению и дупликации митохондриальной ДНК.
Хотя болезнь и является генетической, обычно мутация появляется спонтанно, и больной малыш рождается у абсолютно здоровых родителей. Иногда отмечают зависимость между наличием офтальмопатии у матери и развитием синдрома Пирсона у ее ребенка.
Дефекты ДНК удается выявить в костном мозге, ациноцитах поджелудочной железы, а также в органах, которые не являются главными мишенями болезни – почках, сердечной мышце, гепатоцитах. С другой стороны, у некоторых пациентов при наличии типичной клинической и лабораторной картины так и не удается зарегистрировать изменения в митохондриальной ДНК.
У больных детей происходит накопление железа в печени, склероз клубочков почек, образование кист. В некоторых случаях развивается фиброз миокарда, что приводит к сердечной недостаточности.
Поджелудочная железа выделяет недостаточное количество липазы, амилазы и бикарбонатов у всех пациентов с болезнью Пирсона. Синдром проявляется атрофией ткани железы и ее последующим фиброзом.
Методы диагностики
С уверенностью поставить диагноз могут только врачи-генетики после исследования митохондриальной ДНК. Также важную роль играет обычный анализ периферической крови: выявляют макроцитарную анемию тяжелой степени, нейтропению и тромбоцитопению. Примечательным является отсутствие эффекта от лечения анемии «Цианкобаламином» и препаратами железа.
Благодаря пункции костного мозга можно увидеть уменьшение общего количества клеток, наличие вакуолей в эритробластах и появление кольцевидных сидеробластов.

Симптомы болезни
Уже с первых дней жизни ребенка можно заподозрить синдром Пирсона. Симптомы болезни дебютируют у младенцев в виде злокачественной анемии и инсулинзависимого сахарного диабета. Наблюдаются бледность кожи, сонливость, вялость, диарея, периодическая рвота, ребенок плохо прибавляет в весе. Пища почти не усваивается, характерна стеаторея. Возникают симптомы сахарного диабета, повышается уровень глюкозы в крови, и появляется склонность к ацидозу. Возможно развитие печеночной, почечной и сердечной недостаточности.
Иногда кроме анемии возникает панцитопения (дефицит не только эритроцитов, но и тромбоцитов и лейкоцитов), что будет проявляться склонностью к кровотечениям и частому присоединению инфекций.

Лечение и прогноз
К сожалению, врачи до сих пор не знают, как победить синдром Пирсона. Лечение его неспецифическое и дает лишь кратковременные результаты.
Анемия не поддается стандартной терапии и требует частого переливания крови. Для улучшения функции поджелудочной железы назначают прием ферментов, а для коррекции метаболических нарушений – инфузионную терапию. В редких случаях проводят трансплантацию костного мозга.
Пирсона синдром имеет неблагоприятный прогноз: дети отстают в физическом развитии, большинство погибает до двух лет. В единичных случаях пациенты живут дольше благодаря эффективной поддерживающей терапии, однако в более старшем возрасте болезнь приводит к мышечной атрофии, характерной для синдрома Кернса-Сейра.
Тяжесть течения болезни во многом зависит от степени поражения ДНК.
Источник