Синдром пены шокейра что такое

Синдром Пена-Шокейра. Пентада Кантрелла
Синдром Пена-Шокейра (Реnа-Shokeir) является наследственным заболеванием, которое впервые было описано S.D.J. Реnа и М.Н.К. Shokeir в 1974 году. Этот синдром характеризуется артрогрипозом и дисморфическими признаками, возникающими вследствие акинезии плода.
Синонимы. Секвенция акинезии/гипокинезии плода и секвенция фетальных акинетических деформаций.
Этиология. Аутосомно-рецессивная форма наследования заболевания является наиболее распространенной. Имеется несколько описаний необычных проявлений, которые указывают на возможность гетерогенной этиологии.
Риск рецидива. Прогноз риска рецидива бывает неопределен в связи с мультифакториальностью этиологии. В большинстве случаев он находится в пределах от 0 до 25%. Распространенность. Неизвестна.
Патофизиология. Активное шевеление плода начинается с середины первого триместра беременности и имеет основное значение для нормального развития суставов и прилегающих тканей. Отсутствие или уменьшение внутриутробной двигательной активности приводит к тугоподвижности суставов, формированию птеригумов и нарушениям нейромышечных функций со снижением глотательных движений у плода, что вызывает гипоплазию легких и многоводие.
Диагностика. Выявление сочетания патологического положения конечностей, ограниченности движений плода со сниженной или отсутствующей реакцией на акустическую стимуляцию, задержки внутриутробного развития, многоводия и гипоплазии легких позволяет установить диагноз. Также могут выявляться низкое расположение и аномалии ушных раковин, гипертелоризм, короткая шея, расщелина неба, отек мягких тканей головы, деформации грудной клетки, камподактилия и микрогнати. Реже описываемые аномалии при синдроме ПенаШокейра (Pena-Shokeir) включают диафрагмальную грыжу, гастрошизис и микроцефалию.
Дифференциальный диагноз. Трисомия 18 может сопровождаться наличием признаков аналогичных тем, которые обнаруживаются при синдроме Пена-Шокейра (Pena-Shokeir), особенно в отношении черепно-лицевых и внутригрудных аномалий, а также патологии конечностей. Установить диагноз позволяет проведение кариотипирования. Похожее сочетание признаков, за исключением гипоплазии легких, также наблюдается при нелетальных формах артрогрипоза.
Прогноз. Синдром Пена-Шокейра (Pena-Shokeir) является летальным заболеванием. Значительное число пораженных плодов рождается преждевременно. В 30% срочных родов наблюдается мертворождение. Среди выживших большинство детей погибает в течение первых недель жизни. Основной причиной смертности является легочная недостаточность.
Акушерская тактика. Стандартная акушерская тактика ведения беременности изменяется только при возникновении показаний со стороны матери.
Пентада Кантрелла
Данное врожденное заболевание характеризуется двумя основными дефектами: эктопией сердца и дефектом передней брюшной стенки (чаще всего наблюдается омфалоцеле, но может встречаться и гастрошизис) в сочетании с нарушением развития трех, связанных между собою структур: дистального отдела грудины, передней части диафрагмы и диафрагмального отдела перикарда. Имеются сообщения о вариантах классической формы, которые характеризуются неполным проявлением данного синдрома.
Синонимы. Торако-абдоминальная эктопия сердца, эктопия сердца, синдром Кантрелла-Хеллера-Равича (Cantrell-Heller-Ravich); синдром пентады и перитонеоперикардиальная диафрагмальная грыжа.
Распространенность. Встречается очень редко. В литературе имеются сообщения приблизительно о 90 клинических наблюдениях и еще меньшем их числе с полным подтверждением имевшегося синдрома.
Этиология. Неизвестна. Иногда может сочетаться с хромосомными аномалиями.
Риск рецидива. Неизвестен. Данных о рецидивах не имеется. Опубликовано сообщение об одном случае конкордантного развития синдрома у монозиготных близнецов.
Диагностика. Наличие полной формы дизрупции порока развития передних стенок грудной клетки и брюшной полости, которая сопровождается пятью признаками, описанными J.R. Cantrell et al., является классической формой пентады и может быть выявлена при ультразвуковом исследовании начиная с середины второго триместра. Обнаружение сокращающегося вне пределов грудной клетки сердца в сочетании с омфалоцеле позволяет установить диагноз пентады. Для исключения хромосомных аномалий рекомендуется проведение кариотипирования. Вследствие компрессии органов как грудной клетки, так и брюшной полости могут возникать асцит и гидроторакс.
Генетические нарушения. Неизвестны.
Патогенез. Остановка развития сегмента латеральной мезенхимы в период от 14-го до 18-го дня после зачатия приводит к незакрытию брюшной стенки и неполному слиянию наружных первичных тяжей.
Сочетанные аномалии. Является правилом наличие внутрисердечных аномалий (таких, как тетрада Фалло (Fallot), дефекты межжелудочковой перегородки). Другие нарушения включают в себя черепно-лицевые аномалии, гидроцефалию, анэнцефалию, незавершенный поворот толстой кишки, косолапость и хромосомные аберрации.
Дифференциальный диагноз. Изолированная торакальная эктопия сердца, эктопия сердца в сочетании с синдромом амниотических перетяжек, аномалия развития стебля тела, изолированное омфалоцеле, синдром Беквита-Видемана (Beckwith-Wiedeman) и хромосомные аномалии.
Прогноз. Выживание является исключительным случаем и зависит от размера дефекта абдоминальной стенки, степени тяжести поражения сердца и наличия сочетанных аномалий. В редких случаях при наличии легких форм возможно проведение хирургической коррекции пороков. В тех ситуациях, когда имеется полная экструзия сердца и органов брюшной полости, прогноз исключительно неблагоприятный.
Акушерская тактика. Родоразрешение рекомендуется проводить в специализированных перинатальных медицинских центрах.
— Также рекомендуем «Синдром Пфайффера. Диагностика и прогноз синдрома Пфайффера»
Оглавление темы «Врожденные синдромы плода»:
1. Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
2. Синдром летального птеригума. Лиссэнцефалия I типа
3. Синдром Мекеля. Диагностика и прогноз при синдроме Мекеля
4. Мезомелическая дисплазия. Диагностика и прогноз мезомелической дисплазии
5. Микроцефалический первичный нанизм. Синдром Неу-Лаксова
6. Синдром Нунан у плода. Секвенция маловодия
7. Несовершенный остеогенез. Диагностика и прогноз несовершенного остеогенеза у плода
8. Синдром Пена-Шокейра. Пентада Кантрелла
9. Синдром Пфайффера. Диагностика и прогноз синдрома Пфайффера
10. Синдром Поланда. Секвенция Поттер и синдром prune-belly
Источник
(описан S. D. J. Pena и M. N. K. Schokeir; синонимы – цереброокулофацио-скелетный синдром, COFS-синдром) – наследственное заболевание с преимущественным поражением мозга, глаз, лица и скелета. Характерны выраженная пренатальная гипоплазия; мышечная гипотония; микроцефалия и гидроцефалия, агенезия мозолистого тела; гипоплазия, гиппокампа верхних теменных извилин, микрогирия базиллярных поверхностей; атрофия зрительных нервов; микрофтальмия, катаракта, блефарофимоз (короткая и узкая глазная щель); лицевые аномалии – скошенный лоб, седловидный нос, большие низко расположенные ушные раковины, тонкие губы, малые размеры верхней челюсти и ее смещение назад; аномалии скелета – кифосколиоз, сгибательные контрактуры крупных суставов, «стопа-качалка», вывих тазобедренных суставов или дисплазия вертлужной впадины, узкий таз и др.; пороки внутренних органов – чаще почек (агенезия или гипоплазия, подковообразная почка); отставание физического и психомоторного развития. Тип наследования – аутосомно-рецессивный. Лечение симптоматическое.
S. D. J. Pena, M. N. K. Schokeir. Autosomal recessive cerebro-oculo-skeletal (COFS) syndrome. Clinical genetics, 1974; 5: 285–295.
Просмотров: 6345
Категория: Словари и энциклопедии » Психология »
Другие новости по теме:
ПЕНЫ – ШОКЕЙРА СИНДРОМ
—
Код для вставки на сайт или в блог:
Код для вставки в форум (BBCode):
Прямая ссылка на эту публикацию:
Данный материал НЕ НАРУШАЕТ авторские права никаких физических или юридических лиц.
Если это не так — свяжитесь с администрацией сайта.
Материал будет немедленно удален.
Электронная версия этой публикации предоставляется только в ознакомительных целях.
Для дальнейшего её использования Вам необходимо будет
приобрести бумажный (электронный, аудио) вариант у правообладателей.
На сайте «Глубинная психология: учения и методики» представлены статьи, направления, методики по психологии, психоанализу, психотерапии, психодиагностике, судьбоанализу, психологическому консультированию; игры и упражнения для тренингов; биографии великих людей; притчи и сказки; пословицы и поговорки; а также словари и энциклопедии по психологии, медицине, философии, социологии, религии, педагогике. Все книги (аудиокниги), находящиеся на нашем сайте, Вы можете скачать бесплатно без всяких платных смс и даже без регистрации. Все словарные статьи и труды великих авторов можно читать онлайн.
Источник
Что такое синдром Коккейна?
Синдром Коккейна (СК) — редкая форма карликовости. Это наследственное заболевание, диагноз которого зависит от наличия трех признаков (1) замедления роста, т.е. низкого роста, (2) ненормальной чувствительности к свету (светочувствительность) и (3) преждевременного старения (прогерия).
При классической форме синдрома Коккейна (СК I типа) симптомы прогрессируют и, как правило, проявляются после одного года. Раннее начало или врожденная форма синдрома Коккейна (СК II типа) проявляется при рождении (врожденный). Существует третья форма, известная как синдром Коккейна III типа (СК III типа), которая проявляется позднее при развитии ребенка и, как правило, является более легкой формой заболевания. Четвертая форма; в настоящее время названная комплекс пигментной ксеродермы/синдрома Коккейна (ПК/СК комплекс), сочетает в себе признаки обоих этих расстройств.
Признаки и симптомы
Симптомы всех форм синдрома Коккейна схожи. Различные типы болезни определяются возрастом начала.
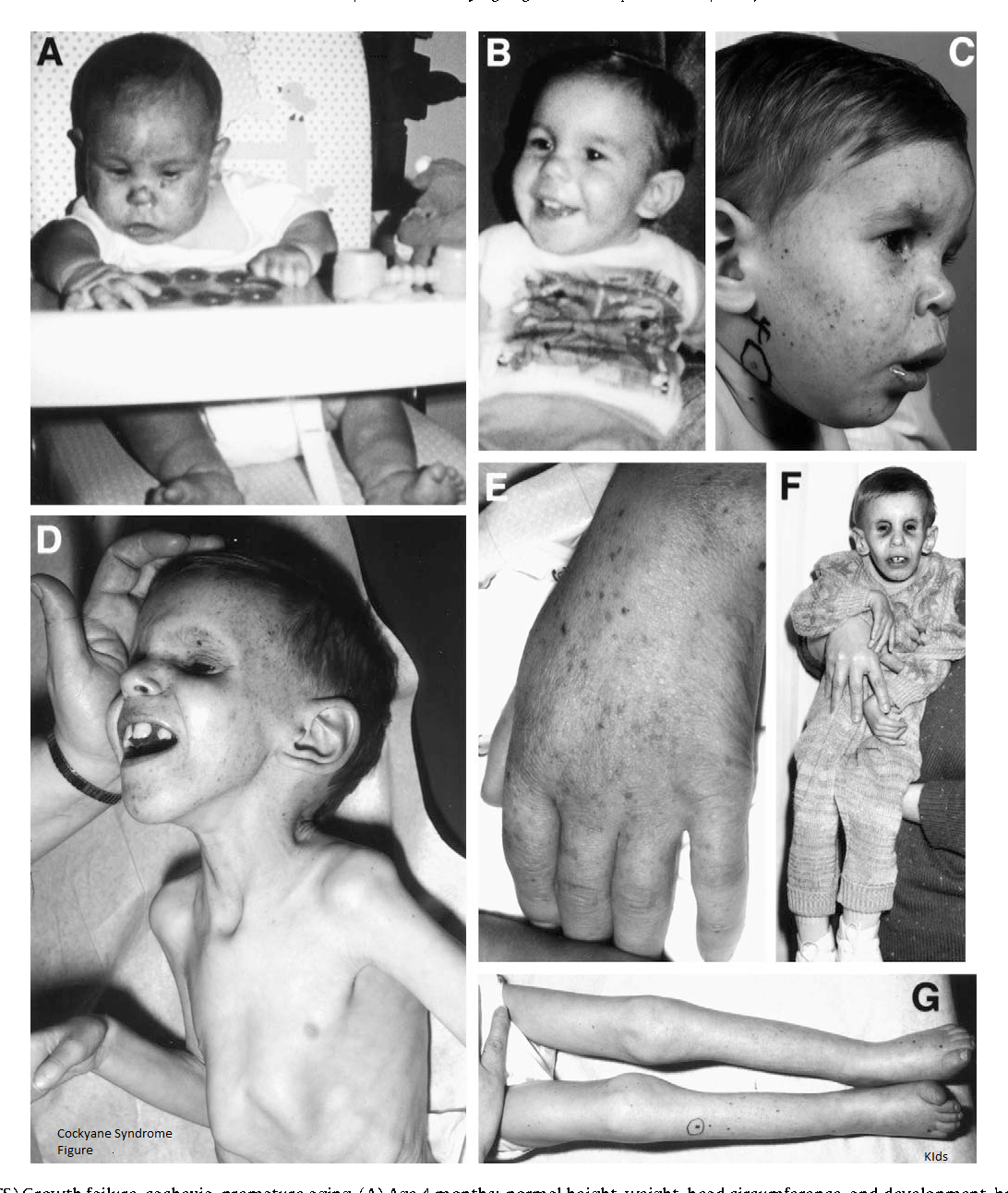
СК I типа, классическая форма, характеризуется нормальным появлением новорожденного, симптомы могут проявляться только после первого года. Рост и вес, а также другие показатели роста и размера находятся в пределах 5-го процентиля. Со временем ухудшается зрение, слух и функционирование нервной системы (центральной и периферической), что может привести к серьезной инвалидности.
Немногочисленные случаи врожденного СК II типа, о которых сообщалось, характеризуются очевидной недостаточностью роста при рождении, а также небольшим или отсутствием неврологического развития после рождения. Серьезные нарушения зрения (катаракта и другие структурные нарушения глаза) обычно присутствуют при рождении. Также встречаются ранние скелетные аберрации. Вероятно, что СК II типа включает некоторых пациентов, ранее диагностированных с цереброокулофациоскелетным синдромом и синдромом Пены-Шокейра II типа из-за идентификации общего дефекта гена у этих пациентов.
III тип СК еще более редок и характеризуется в основном нормальным ростом и умственным развитием в первые годы, но прерывается поздним началом типичных симптомов синдрома Коккейна.
ПК/СК комплекс является наиболее редкой формой и включает в себя признаки обоих заболеваний. Широко распространенные веснушки и ранний рак кожи типичны для пигментной ксеродермии, а невысокий рост, умственная отсталость и недостаточное сексуальное развитие типичны для СК.
Основные характеристики синдрома Коккейна включают:
- задержку нормального роста (карликовость) в позднем детстве;
- чрезвычайную чувствительность к свету (светочувствительность);
- преждевременное старение (прогероид).
Кожа кажется сморщенной и состарившейся, особенно на лице, руках и ногах, из-за потери жира под кожей (подкожная жировая ткань). Дети с этим заболеванием могут иметь повышенную пигментацию кожи.
Дети с синдромом Коккейна имеют необычные физические особенности, включая:
- ненормально маленькую голову (микроцефалия)
- необычно тонкий нос
- «впалый» или утонувший вид в глаз
- большие деформированные уши
- выступание вперед челюстей (прогнатизм).
Может быть необычное количество кариеса из-за неправильного расположения зубов. Пострадавшие люди обычно имеют необычно длинные руки и ноги пропорционально размеру их тела. Суставы также могут быть аномально большими и оставаться в фиксированном положении (согнутыми), а позвоночник может быть изогнут наружу, если смотреть сбоку (кифоз). Другие особенности синдрома Коккейна могут включать снижение потливости (гипогидроз), отсутствие слез в глазах, преждевременную седину.
Другие симптомы синдрома Коккейна могут включать аномальный синий оттенок кожи (цианоз) на руках и ногах, который также может быть холодным на ощупь. Неврологические симптомы могут включать ритмичные, дрожащие движения (тремор), неустойчивую походку (атаксию) и/или неспособность координировать движение. Больные дети могут испытывать различные степени умственной отсталости, частичной потери слуха и/или прогрессирующей потери ранее приобретенных интеллектуальных способностей.
Симптомы синдрома Коккейна, которые влияют на глаза (глазные признаки), могут включать:
- прогрессирующее помутнение хрусталика глаз (катаракта);
- потерю зрения из-за истощения нервных волокон в глазах (атрофия зрительного нерва);
- дегенерацию сетчатки;
- пигментацию сетчатки.
У некоторых людей с синдромом Коккейна также может быть аномально высокое кровяное давление (артериальная гипертензия), увеличение печени (гепатомегалия) и/или преждевременное накопление жировых бляшек на стенках артерий вокруг сердца (атеросклеротическая болезнь). Взрослые с этим расстройством могут быть сексуально недоразвиты.
Причины (этиология)
На молекулярном уровне СК вызван дефектом одного из генов, участвующих в нормальной репарации ДНК, которая была повреждена ультрафиолетом. Это естественная защита организма от солнечных ожогов. Воздействие ультрафиолетового компонента солнечного света повреждает ДНК, но клетка больше не способна восстанавливать поврежденную ДНК по мере ее образования и накопления в клетке.
Вполне вероятно или, по крайней мере, подозревается, что некоторые из генов, которые вызывают СК, также участвуют в синтезе белка, и что другие признаки СК являются результатом продукции и накопления аномальных белков в клетке.
Ген, ответственный за СК I типа, картирован в хромосоме 5 и называется ERCC8. Ген II типа СК был картирован в хромосомном локусе 10q11 и называется ERCC6. Мутации в ERCC6 составляют около 75% случаев, в то время как мутации в ERCC8 вызывают около 25% случаев.
Синдром Коккейна наследуется как аутосомно-рецессивный генетический признак. Человеческие черты, включая классические генетические заболевания, определяются двумя генами, один из которых получен от отца, а другой — от матери. Рецессивные расстройства возникают, когда человек наследует один и тот же аномальный ген по одному признаку от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно не проявлять симптомов. Риск для двух родителей-носителей, передать оба дефектных гена и, следовательно, иметь больного ребенка, составляет 25% с каждой беременностью. Риск иметь ребенка-носителя как и родители, составляет 50% с каждой беременностью. Риск для ребенка получить нормальные гены от обоих родителей и быть генетически нормальным для этой конкретной черты составляет 25%.
Затронутые группы населения
Синдром Коккейна встречается очень редко и поражает мужчин и женщин в равных количествах. Нет признаков этнической или расовой принадлежности. Заболеваемость СК составляет менее 1 случая на 250 000 живорождений. По состоянию на 1992 г. в литературе было зарегистрировано около 140 случаев СК.
Связанные расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Коккейна. Сравнения могут быть полезны для дифференциальной диагностики:
- Синдром Хатчинсона-Гилфорда (прогерия) — очень редкое заболевание детского возраста, характеризующееся преждевременным старением, невысоким ростом и необычными чертами лица. Первичные симптомы расстройства связаны с процессом старения, включая седые волосы, морщинистую кожу, артрит и болезни сердца. Новорожденные дети с синдромом Хатчинсона-Гилфорда имеют нормальную массу тела при рождении, но в течение первого года жизни происходят глубокие нарушения роста. Приблизительно в 10 лет большинство детей с синдромом Хатчинсона-Гилфорда достигают роста в среднем как трехлетние дети и имеют много проблем со здоровьем как пожилые люди.
- Синдром Секкеля (или сочетание карликовости с «птицеголовостью») — редкое генетическое заболевание, характеризующиеся недостаточностью роста плода и младенцев, умственной отсталостью и типичными чертами лица. Физические характеристики включают аномально маленькие челюсти (микрогнатия) и голову (микроцефалия), а также выпуклость средней части лица и нос в форме клюва. Уши обычно посажены низко и деформированы, а глаза необычно большие. Также может присутствовать аномальная кривизна позвоночника (сколиоз) и недоразвитие наружных половых органов.
- Карликовость Ларона — редкое генетическое заболевание, характеризующееся небольшим ростом, необычными чертами лица и аномально высоким уровнем гормона роста в крови. Дети с этим расстройством вырабатывают достаточное количество этого гормона, но их организм не может правильно использовать его из-за отсутствия рецепторов гормона роста. Младенцы с карликовостью Ларона имеют серьезную задержку роста, задержки в появлении зубов, непропорциональный рост между макушкой головы и челюстями, плоский широкий нос и/или глубоко посаженные глаза.
Методы диагностики
Диагностика основана на обнаружении специфического дефекта TCR, который можно идентифицировать с помощью радиоактивного анализа в культивируемых фибробластах, который измеряет восстановление синтеза РНК после УФ-облучения. Этот тест репарации ДНК является решающим инструментом для диагностики СК. Специализированное визуальное тестирование (МРТ) может продемонстрировать потерю жирового покрытия (демиелинизация) на некоторых нервных волокнах мозга.
Стандартные методы лечения
Лечение синдрома Коккейна носит исключительно поддерживающий и симптоматический характер, и включает физиотерапию, защиту от солнца, ношение слухового аппарата и часто трубчатое питание или гастростомию.
Прогноз
При СК I типа смерть наступает до конца второго десятилетия в результате прогрессирующей неврологической дегенерации. Пациенты с типом II имеют более тяжелый прогноз (в среднем живут 2-7 лет), тогда как пациенты с типом III живут в зрелом возрасте, поскольку III тип протекает со слабыми симптомами. Люди с III типом синдрома Коккейна живут в зрелом возрасте со средней продолжительностью жизни 40-50 лет.
Источник