Синдром нагера до и после операции

Главная » Болезни и мутации
27.06.2019
: 17929
: 0
операция, хирургия, лицо, аномалия, Синдром Нагера
![]()
Если с вами произошел необычный случай, вы увидели странное существо или непонятное явление, вам приснился необычный сон, вы увидели в небе НЛО или стали жертвой похищения пришельцев, вы можете прислать нам свою историю и она будет опубликована на нашем сайте
===>
Прислать историю
.
6-летняя Дарина Шпенглер из Красноярского края родилась с аномалией, из-за которой у нее отсутствуют губы и подбородок. В целом вся нижняя часть лица у нее не развита. Диагноз девочке официально никогда не был поставлен, но предполагают, что у нее редкий врожденный Синдром Нагера.
Мать ничего не знала об этой аномалии до родов, а когда Дарина родилась, медсестры долго не показывали ребенка матери — Елене Шпенглер. Когда же они наконец принесли ей девочку, то мать упала в обморок, едва взглянув на нее.
Отец же Дарины был совершенно невозмутим, когда увидел ребенка. «Его лицо нисколько не изменилось», — рассказывает Елена.

Когда врачи стали рекомендовать Елене отказаться от ребенка, мать не стала их слушать. Впоследствии из-за состояния Дарины родители девочки разругались с большинством своих родственников (те пеняли им, что не оставили ребенка в больнице) и уехали из деревни, где жили.
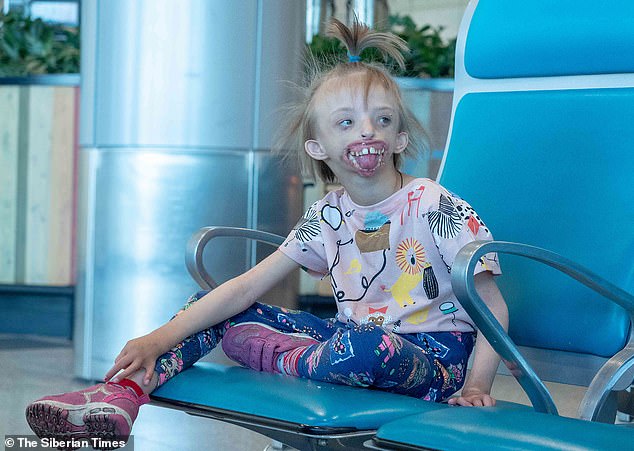
Когда Дарина была младше она много плакала, потому что ткани вокруг челюстей постоянно кровоточили и болели.

Несколько лет родители девочки пытались найти помощь у российских хирургов, однако им никто не смог помочь. Надежда проснулась, когда появился шанс поехать в Великобританию в знаменитую лондонскую детскую больницу на Грейт Ормонд -стрит.
Из-за открытых челюстей Дарине очень сложно есть, поэтому в свои шесть лет она весит ниже нормы и на вид ей не дашь больше четырех. После операции, которая закроет челюсти, она сможет наконец питаться как все люди.
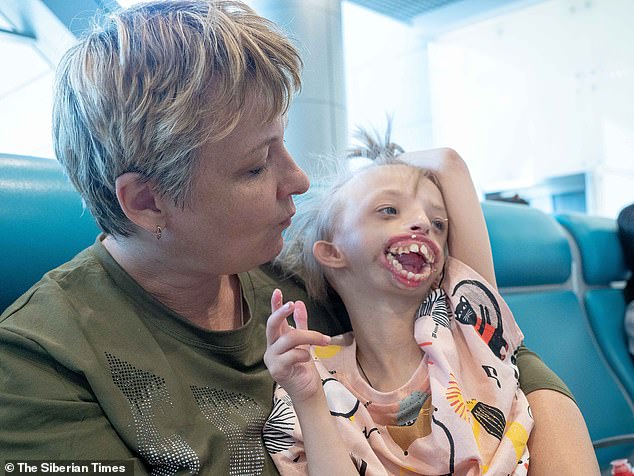
Но больше всего родители девочки надеются на то, что во время операции ей исправят и голосовые связки, ведь Дарина пока не может произнести ни слова.
Внешность Дарины шокирует многих неподготовленных людей. Прохожие на улицах смотрят ей вслед, а некоторые даже советуют родителям девочки надевать на Дарину маску. Из-за этого друзей среди детей у Дарины нету, дети боятся играть с ней.

Однажды родителей девочки даже обвинили в том, что они сами нанесли ей эти раны. Им звонила полиция и требовала рассказать, что произошло. Лишь после всех объяснений полиция перестала их беспокоить.

Дарина имеет нормальное умственное развитие и уже понимает, что с ней что-то не так. Родители переживают из-за этого, но отказываются прятать девочку от общества и тем более надевать на нее маску. Они принимают ее такой, какая она есть, даже несмотря на то, что девочку из-за ее внешности не приняли ни в один детский сад. Пару раз в неделю на дом к Дарине приходят учителя и учат ее читать и писать.
Дарина и ее мама Елена уже прилетели в Лондон и сейчас готовятся к операции. Стоимость операции равна 85 тысячам долларов и оплачивается российскими благотворителями через общество Русфонд.
Синдром Нагера влияет на лицевые кости и он настолько редок, что в мире зафиксировано лишь около 100 таких случаев.
Источник
Печально наклоненные вниз наружные уголки глаз, большой нос и миниатюрный подбородок — людей с синдромом Тричера-Коллинза можно узнать в лицо. Это аутосомно-доминантное заболевание встречается у 1 из 50 000 младенцев. Значит, в России ежегодно рождается около 30-40 таких детей. Немного в масштабах огромной страны и невероятно много, когда речь идет о человеческих судьбах.
К сожалению, в российской практике жизнь людей с синдромом Тричера-Коллинза осложняется большими немедицинскими проблемами — нехваткой специалистов на местах и негативным отношением окружающих. Но если второй момент можно сгладить, окружив ребенка заботой в семье, то поиск адекватного лечения порой занимает слишком много времени.
Из-за отсутствия необходимой информации и адекватного лечения родители упускают шанс скомпенсировать заболевание, ребенок начинает отставать в развитии. А ведь эти дети имеют сохранный интеллект. Они способны к обучению и к активной жизни. Доказательством тому служит и инструктор по фитнесу из Великобритании Йоно Ланкастер, который сейчас сам помогает детям с таким же заболеванием, и фотомодель Элисон Мидстокк из Нью Йорка.
Мы расскажем, что это за синдром, как его диагностируют, как и где лечить пациентов разных возрастов.
Синдром Тричера-Коллинза — только факты
Синдром Тричера-Коллинза (Treacher Collins syndrome, TCS или челюстно-лицевой дизостоз) вызывается генетической мутацией, чаще всего в гене TC0F1, а также в генах POLR1C и POLR1D. Примерно в 40% случаев мутация передается наследственным путем от одного из родителей к ребенку, а в 60% случаев мутация является спонтанной (de novo). У родителя с синдромом Тричера Коллинза существует 50%-ная вероятность рождения ребенка с этим синдромом.
При этом заболевании некоторые кости и ткани лица не полностью сформированы, причем проявления могут варьироваться от почти незаметных признаков до крайне тяжелых форм.
- Люди с незначительными дефектами иногда даже не подозревают о наличии генетических отклонений.
- Средняя степень характеризуется специфической формой лица, словно «смятой» вглубь, маленькой нижней челюстью и подбородком. Частым признаком является недоразвитие ушных раковин и дефекты слуховых проходов, а также дефекты неба.
- Нарушения сильной степени требуют проведения десятков пластических операций, иногда — хирургии носовых проходов, различных мероприятий по восстановлению слуха.
Из-за чересчур маленьких челюсти и подбородка у пациентов возникают такие проблемы с дыханием, которые требуют установки трахеостомы. Синдром Тричера-Коллинза нередко сопровождается двусторонней потерей слуха. Дефекты развития век могут включать невозможность полностью закрыть глаза. А вот умственное развитие детей с синдромом Тричера Коллинза обычно нормальное, если у них нет серьезных проблем с дыханием.
Основные проявления синдрома Тричера-Коллинза:
- Очень маленькие челюсть и подбородок (микрогнатия)
- Выемка в области нижнего века (колобома века) и плохо сформированные нижние веки с редкими ресницами
- Отсутствующие, маленькие или необычно сформированные уши (анотия или микротия)
- Потеря слуха
- Узкий или отсутствующий наружный слуховой проход (атрезия)
- Дефекты слуховых косточек
- Недоразвитые или отсутствующие скуловые и орбитальные кости лица
- Наклоненные вниз наружные углы глаз
- Большой нос
- Суженные, полностью или частично заблокированные носовые проходы
- Волчья пасть
Все эти черты могут вызывать проблемы со зрением, глотанием, дыханием и слухом.
Вполне возможно, что один из родителей ребенка с тяжелыми проявлениями синдрома Тричера Коллинза может тоже быть носителем этого нарушения и не знать этого, так как в его случае проявление симптомов может быть очень незначительным и незаметным.
Диагностика синдрома Тричера Коллинза
В большинстве случаев синдрома Тричера Коллинза врачи ставят диагноз вскоре после рождения или в течение первого года жизни. Однако диагноз может быть установлен и гораздо позже в случае очень незначительного проявления симптомов.
Врачи, знакомые с этим синдромом, часто могут поставить диагноз на основе того, как выглядит лицо пациента. Иногда требуется трехмерная (3D) компьютерная томография, чтобы выявить все детали строения лица. Диагноз подтверждают с помощью генетического анализа.
В Детской больнице Цинциннати (Cincinnati Children’s Hospital) действует единственный в США мультидисциплинарный Центр синдрома Тричера Коллинза. Здесь работают эксперты разных специальностей, имеющие опыт лечения всех проявлений этого синдрома.
Лечение синдрома Тричера Коллинза
Лечение подразумевает комплексное устранение симптомов и их последствий. Специалисты Центра синдрома Тричера Коллинза создают индивидуальные планы лечения, отвечающие потребностям каждого ребенка, координируют это лечение и подключают другие отделения больницы, такие как:
- Генетика человека
- Пульмонология
- Пластическая хирургия
- Отоларингология
- Неонатология
- Аудиология
- Патологии развития речи
- Специалисты по кормлению
- Центр верхних дыхательных путей
Например, Отделение генетики человека играет важную роль в лечении и ведении пациентов с этим диагнозом. По итогам комплексного генетического обследования специалисты спрогнозируют риск наследования заболевания другими детьми в семье или потомством пациента и репродуктивные варианты. В некоторых случаях возможно пренатальное обследование.
Если ребенок рождается с серьезными проблемами с дыханием, его обследует специальная группа специалистов вскоре после рождения. Цель этого обследования – как можно скорее поставить диагноз и начать лечение.
Выбор методов лечения основывается на возрастном подходе:
Младенчество. Главное в этом возрасте — убедиться, что дыхательные пути младенца стабильно функционируют. Иначе нехватка кислорода скажется на умственном развитии. В этот период могут сделать операцию по удлинению нижней челюсти, установить трахеостому. Проблемы с кормлением также нередко требуют немедленного решения. Если есть проблемы со слухом, возможна установка кохлеарных имплантов.
Детство. По мере того, как ребенок растет и начинает ходить в школу, врачи уделяют больше внимания внешнему виду лица. Детям 6-8 лет может быть сделана еще одна операция по удлинению нижней челюсти, если у них до сих пор есть проблемы с апноэ во сне. Среди других операций самые частые — исправление век и реконструкция ушей.
Подростковый возраст. Подросткам могут потребоваться дополнительные хирургические процедуры для исправления дефектов лица в зависимости от того, как их лицевые кости и ткани росли и развивались в детстве. Обычно это операции на верхнюю или нижнюю челюсть, пересадку кости и исправление формы носа. В это же время следует обратиться к услугам ортодонтов.
Во многих случаях специалисты Центра ведут пациентов с синдромом Тричера-Коллинза до 22 лет, после чего, если есть необходимость, помогают им перейти под наблюдение взрослых специалистов.
Результаты лечения
Одна из основных целей врачей Центра, имеющих большой опыт лечения детей с синдромом Тричера Коллинза,— поддерживать их дыхательные пути в стабильном состоянии, когда только возможно избегая установки трахеостомы. Они также стремятся исключить длительное использование приборов для кормления (таких как назогастральный зонд) у детей, которым сделали операцию по удлинению челюсти.
Научный прогресс и развитие технологий, таких как кохлеарные импланты и операции по удлинению челюсти, позволяют многим детям с синдромом Тричера-Коллинза вести нормальную жизнь.
Далекая перспектива зависит от того, насколько сильны проявления синдрома. Легкие случаи могут требовать минимального лечения, в то время как пациенты с серьезными проблемами с дыханием могут всю жизнь нуждаться в трахеостоме.
Повторимся, этот синдром не влияет на способности детей к обучению и к активной жизни.
Некоторые симптомы синдрома Тричера-Коллинза сходны с симптомами других заболеваний, относящихся в группе нижнечелюстно-лицевых дизостозов. Еще одна группа заболеваний, акрофациальные дизостозы (такие как синдром Нагера, синдром Миллера и др.), также имеет сходные симптомы, но включает и дефекты развития конечностей. Подход к лечению этих заболеваний одинаков, и специалисты Центра Тричера-Коллинза располагают опытом и знаниями в лечении всех этих синдромов.
Здесь маленький пациент получает всю необходимую помощь в одном месте. Эта модель устраняет стресс и разочарование при планировании ухода в нескольких учреждениях и позволяет получить наилучшие возможные результаты.
Источник
Эта статья создана пользователем Онедио. Со стороны редакции изменений не было. Вы тоже можете создавать свои статьи на нашем сайте.
Onedio избранное > Интересно-16 января 2018, 09:57‘ добавлено,
17 января 2018, 11:23‘ обновлено
Наверняка у вас тоже есть недостатки, которых вы стесняетесь и которые всячески стараетесь исправить… Не переживайте слишком сильно по этому поводу, ведь у некоторых людей такие недостатки имеют гораздо более серьёзное происхождение (да и их и недостатками уже назвать нельзя, это самые что ни на есть настоящие уродства). Посмотрим?
Источник: https://list25.com/25-people-with-strang…
1. Семья Улас
list25.com
Эта семья из Турции всё время передвигается на четырёх конечностях, а всё из-за редкого вида инвалидности. Людям просто не хватает равновесия, чтобы держаться на двух ногах.
2. Хосе Местре
Показать изображение
list25.com
Опухоль весом в 5.5 кг буквально целиком съела лицо португальца Хосе Местре. Такое уродство вызвало сильный рост кровеносных сосудов, который ослепил мужчину, добрался до его рта (затрудняя речь и мешая есть). Хирург из Чикаго взялся за сложнейшую операцию по удалению опухоли, и ему это успешно удалось.
3. Неизвестный
list25.com
Звучит иронично, но у людей действительно могут расти рога. Всё из-за излишнего количества кератина, который вырабатывается кожей. К счастью, такие рога можно удалить хирургическим путём…
4. Бри Уолкер
Телеведущая из Лос-Анджелеса живёт с врождённым пороком под названием эктродактилия («клешнеобразная кисть»). Порок заключается в недоразвитии одного или нескольких пальцах на кистях или стопах.
5. Хавьер Ботет
Этого мексиканского актёра также называют ходячим спецэффектом, ведь именно его зовут сниматься во многих ужастиках. А всё из-за врождённого генетического заболевания, которое называется «болезнь Марфана» или «Синдром Марфана». Люди с таким заболеванием отличаются неестественной худобой, высоким ростом, а также удлинёнными конечностями и пальцами.
6. Петеро Бьякатонда
list25.com
Петеро родом из небольшого городка в Уганде и страдает от врождённой аномалии развития черепа под названием синдром Аперта. У страдающих этим заболеванием наблюдается отсталость умственного развития, выпуклый лоб, вогнутое лицо, а также деформация челюсти.
7. Руди Сантос
Показать изображение
list25.com
Когда Руди был в утробе матери, у него был брат-близнец, но затем Руди просто его поглотил, в результате чего начал страдать от паразитарной краниопаги (особый тип сращения сиамских близнецов). Сейчас у Руди дополнительная пара ног и рук 
8. Харри Реймонд Истлек
list25.com
Харри страдал от очень редкого недуга под названием прогрессирующая оссифицирующая фибродисплазия (ФОП). При этом у человека соединительные ткани начинают превращаться в кости, в результате чего формируется буквально новая структура скелета.
9. Деде Косвара
Показать изображение
list25.com
Этого индонезийца также называют «человеком-деревом», ведь всё его тело покрывают ужасные наросты в виде бородавок. Иммунная система этого мужчины не может побороть такой активный рост бородавок. К сожалению, в 2016 году мужчина скончался.
10. Дидье Монтальво
list25.com
Этот мальчик из Колумбии страдает заболеванием, из-за которого по всему его телу растут родинки. Одна из них (на спине) стала настолько большой, что напоминает панцирь черепахи. Слава Богу, что британскому хирургу удалось помочь мальчику, удалив родинку, и теперь он живёт нормальной жизнью.
11. Тесса Эванс
list25.com
Девочка страдает от аплазии — врождённый порок, который заключается в отсутствии какой-либо части тела (в данном случае носа). К счастью, таким детям можно прибегать к протезированию, но вот запахов Тесса слышать уже никогда не будет.
12. Дин Эндрюс
list25.com
Дин страдает от редкого генетического дефекта под названием «прогерия». Он заключается в преждевременном старении организма. Сейчас Дину всего 20, но выглядит он на 100 лет!
13. Неизвестная
list25.com
Синдром Тричера Коллинза — это аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией. Оно может повлиять на развитие скул, челюсти, подбородка и ушей человека. В некоторых случаях дефекты можно исправить пластическими операциями.
14. Деклан Хайтон
list25.com
Этот мальчик из Великобритании страдает от врождённого Синдрома Мёбиуса, из-за которого он не может двигать мышцами лица (в том числе глазами).
15. Верн Тройер
list25.com
Этот американский актёр страдает от дисплазии — генетической болезни, которая передалась ему от отца. Поэтому его рост равен всего 80 см.
16. Манар Магед
list25.com
Как и Руди Сантос, Манар Магед страдает от от паразитарной краниопаги. Но хирурги не смогли разделить близнецов, поэтому девочка скончалась.
Эта статья создана пользователем Онедио. Со стороны редакции изменений не было. Вы тоже можете создавать свои статьи на нашем сайте.
Источник
Синдром Тричера Коллинза (англ. Treacher Collins syndrome, TCS, челюстно-лицевой дизостоз) — аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией. Описан английским офтальмологом Эдвардом Тричером Коллинзом в 1900 году[1].
Синдром Тричера Коллинза встречается у 1 из 50 000 младенцев[2]. Типичные клинические признаки: грубый дефект лицевой части черепа, косоглазие, колобомы век; размер рта, подбородка и ушей существенно меньше нормы. В некоторых случаях — ослабление слуха.
Этиология[править | править код]
Причиной заболевания является, чаще всего, нонсенс-мутация (возникновение стоп-кодона) в гене TCOF1, приводящая к гаплонедостаточности.
Ген TCOF1 расположен в локусе 5q32-33, на длинном (q) плече 5-й хромосомы, начинается от 149 737 202-й пары оснований и заканчивается на 149 779 871-й паре оснований[3][4].
Продукт гена TCOF1 — ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. При синдроме Тричера Коллинза развивается состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма[5].
Синдром наследуется по аутосомно-доминантному принципу и характеризуется высокой пенетрантностью. Экспрессивность может быть от умеренной до выраженной, потому тяжесть дефекта отлична у разных пациентов — от почти незаметных признаков до крайне тяжёлых форм. У большинства пациентов слаборазвитые лицевые кости, что приводит к «затонувшему» лицу, крупный нос и очень маленькие челюсти и подбородок (микрогнатия). У некоторых больных присутствует волчья пасть. В тяжелых случаях микрогнатия может вытеснять язык пострадавших новорожденных достаточно, чтобы вызвать преграду ротоглотки и потенциально опасных для жизни заболеваний дыхательных путей. Врождённый порок сердца является необычной особенностью[6].
Лечение[править | править код]
В лечении пациентов, пострадавших от ТКС, применяется междисциплинарный подход, то есть требуются вмешательства различных специалистов. Основные проблемы у пациентов с ТКС — нарушения глотания и проходимости дыхательных путей. Некоторые пациенты нуждаются в трахеостомии. Гастростома может быть необходима для обеспечения адекватного потребления пищи и для защиты дыхательных путей[7]. Вопрос о хирургическом восстановлении структуры лица решается индивидуально и осуществляется по достижении определённого возраста[8].
Потеря слуха при синдроме Тричера Коллинза вызвана деформацией структур в наружном и среднем ухе. Потеря слуха, как правило, двусторонняя. Даже в тех случаях, когда ушные раковины и наружные слуховые проходы не затронуты синдромом, цепи слуховых косточек часто повреждены[9].
Попытки хирургическим путём восстановить наружный слуховой проход для улучшения слуха у детей с ТКС не дали положительных результатов[10]. Предпочтительной оказалась слуховая реабилитация со слуховыми аппаратами костной проводимости[en] (BAHA) или обычными слуховыми аппаратами[11].
В культуре[править | править код]
- «Чудо» — художественный фильм-драма 2017-го года с главным героем с синдромом ТК
Примечания[править | править код]
- ↑ Treacher Collin E, «Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones», 1900, Trans Ophthalmol Soc UK, 20, p. 190—192
- ↑ Chiara Conte, Maria Rosaria D’Apice, Fabrizio Rinaldi, Stefano Gambardella, Federica Sangiuolo and Giuseppe Novelli. Novel mutations of TCOF1 gene in European patients with Treacher Collins syndrome (англ.) // BMC Med Genet. : journal. — 2011. — 27 September (vol. 12). — doi:10.1186/1471-2350-12-125. — PMID 21951868.
- ↑ Dixon M.J., Dixon J., Raskova D., et al. Genetic and physical mapping of the Treacher Collins syndrome locus: refinement of the localization to chromosome 5q32-33.2 (англ.) // Human Molecular Genetics (англ.)русск. : journal. — Oxford University Press, 1993. — Vol. 1, no. 4. — P. 249—253. — doi:10.1093/hmg/1.4.249. — PMID 1303194.
- ↑ Dixon M.J., Dixon J., Houseal T., et al. Narrowing the position of the Treacher Collins syndrome locus to a small interval between three new microsatellite markers at 5q32-33.1 (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 1993. — Vol. 52, no. 5. — P. 907—914. — PMID 8488840.
- ↑ Синдром Тричер Коллинза-Франческетти (мандибулофациальный дизостоз, TCOF) //Центр молекулярной генетики Медико-генетического научного центра РАМН
- ↑ Электронный научный журнал «Медицина и образование в Сибири»
- ↑ Goel L., Bennur S.K., Jambhale S. Treacher Collins syndrome-a challenge for anaesthesiologists (англ.) // Indian Journal of Anaesthesia (англ.)русск. : journal. — 2009. — August (vol. 53, no. 4). — P. 496—500. — PMID 20640217.
- ↑ Evans, Adele Karen; Rahbar, Reza; Rogers, Gary F.; Mulliken, John B.; Volk, Mark S. Robin sequence: A retrospective review of 115 patients (англ.) // International Journal of Pediatric Otorhinolaryngology : journal. — 2006. — 31 May (vol. 70, no. 6). — P. 973—980. — doi:10.1016/j.ijporl.2005.10.016. — PMID 16443284.
- ↑ Argenta, Louis C.; Iacobucci, John J. Treacher Collins Syndrome: Present concepts of the disorder and their surgical correction (англ.) // World Journal of Surgery (англ.)русск. : journal. — 1989. — 30 June (vol. 13, no. 4). — P. 401—409. — doi:10.1007/BF01660753. — PMID 2773500.
- ↑ Marres, HA; Cremers, CW; Marres, E.H. Treacher-Collins syndrome. Management of major and minor anomalies of the ear (англ.) // Revue de laryngologie — otologie — rhinologie : journal. — 1995. — Vol. 116, no. 2. — P. 105—108. — PMID 7569369.
- ↑ Marres, H.A. Hearing loss in the Treacher-Collins syndrome (неопр.) // Advances in oto-rhino-laryngology. — 2002. — Т. 61. — С. 209—215. — doi:10.1159/000066811. — PMID 12408086.
Ссылки[править | править код]
- «Новое лицо Джулианы» — документальный фильм о девочке, родившейся с синдромом Тричера Коллинза тяжёлой формы
Источник