Синдром моносомии по 7 хромосоме

Хромосомные аномалии при миелодиспластическом синдроме — прогноз
Кариотип клеток костного мозга у больных с миелодиспластическими синдромами (МДС) интенсивно изучается на протяжении последних 10—15 лет. Аномальные клоны выявлены до лечения у 30—50 % пациентов, в некоторых сообщениях приведены более высокие показатели — 60—75 %.
Обнаружение клонов клеток с аномальным кариотипом при миелодиспластическом синдроме (МДС) имеет важное теоретическое и клиническое значение, поскольку свидетельствует о принадлежности этой группы заболеваний к новообразованиям.
Цитогенетические изменения весьма разнообразны, спектр их близок к спектру хромосомных аномалий, наблюдаемых при остром нелимфобластном лейкозе, особенно вторичных.
Наиболее характерны моносомии 5 и 7, а также делеции длинного плеча этих хромосом, появление дополнительной хромосомы 8 и делеции длинного плеча хромосомы 20.
Установлено, что частота обнаружения клонов анеуплоидных клеток нарастает по мере прогрессирования болезни: на относительно ранних этапах она составляет 20—30 %, при появлении начальных признаков трансформации в острый лейкоз — до 40—60 %, при трансформации в острый миелобластный лейкоз — 80—90 %.
Транслокации, специфичные для первичных острых нелимфобластных лейкозах, при миелодисплазиях наблюдаются редко. Есть сообщения о повторяющихся транслокациях t(3;3)(q21;q26), t(8;21)(q22;q22) и t(3;21)(q26;q22). Примеры перестроек длинного плеча хромосомы 3 показаны на рисунке.
Частота (в процентах) характерных аномалий кариотипа при различных миелодиспластических синдромах
Основные хромосомные аномалии, характерные для миелодисплазий:
-7 или 7q-
-5 или 5q-
t(1;7)(q10;p10)
del(12)(р12-р13)
t(2;ll)(p13;q23)
del(13)(обязательно с включением 3q14)
t(6;9)(p23;q34)
del(20)(q11ql3)
+8
t(1;3)(p36;q21)
Перечисленные хромосомные аномалии наблюдаются при различных формах миелодисплазий, но частота их несколько различается.
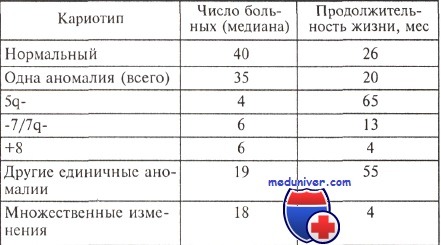
Опыт большинства исследователей свидетельствует о том, что существует корреляция между особенностями кариотипа и продолжительностью жизни больных с миелодиспластическим синдромом (МДС). Относительно благоприятным считается прогноз, если выявлены клоны клеток с единственной перестройкой 5q- или 20q; в то же время при любом варианте миелодиспластического синдрома обнаружение клона с множественными хромосомными аномалиями является крайне неблагоприятным.
Остановимся подробнее на отдельных нарушениях кариотипа, характерных для миелодиспластического синдрома (МДС).
Синдром 5q — рефрактерная сидеробластная анемия у пожилых больных, преимущественно женщин. В новой классификации ВОЗ этот синдром выделен как самостоятельный вариант миелодиспластического синдрома (МДС). Характерна макроцитарная анемия, резистентная к лечению, в костном мозге — признаки миелодисплазии клеток красного ряда и мегакариоцитов. Число тромбоцитов нормально или повышено, в костном мозге наблюдается гиперплазия гиполобулярных микромегакариоцитов. Клиническое течение сравнительно медленное. Трансформации в острый лейкоз наблюдаются приблизительно в 10 % случаев. Синдром впервые описан van den Berghe и соавт. в 1974—1985 гг..
Делеции длинного плеча хромосомы 5 наблюдаются и при других гематологических заболеваниях.
Предполагают, что делетирующийся участок содержит один или более генов-супрессоров. В этом направлении ведутся интенсивные исследования. До настоящего времени не подтверждена важная роль в патогенезе рефрактерной анемии ни одного из изучавшихся кандидатов на роль гена- супрессора.
Продолжительность жизни больных миелодиспластическим синдромом с различными изменениями кариотипа
Синдром моносомии хромосомы 7 встречается преимущественно у мальчиков до 4 лет. Характерна спленомегалия, часто наблюдаются лейкоцитоз с моноцитозом, тромбоцитопения, анемия. Прогноз плохой.
Как отмечалось, утрата одной из хромосом 7-й пары (моносомия 7) наблюдается при самых различных гемобластозах, включая острый нелимфобластный лейкоз, при этом она обычно ассоциирована с неблагоприятным прогнозом.
Делеции короткого плеча хромосомы 17 (17р-) обычно входят в число сложных изменений кариотипа. Как правило, 17р- сочетается с двумя или более хромосомными аномалиями и имеет неблагопрятное прогностическое значение.
В 75 % случаев в присутствии маркера 17р- наблюдается своеобразный дисгранулоцитопоэз в виде псевдопельгеровских гиподольчатых ядер и вакуолизации цитоплазмы. Этот маркер обнаружен не только при миелодисплазиях, но и при самых разнообразных злокачественных новообразованиях, включая солидные опухоли, его присутствие — плохой прогностический признак.
В 1997 г. опубликованы материалы международного совещания, посвященного диагностике и прогнозированию миелодиспластического синдрома. На основании ретроспективной оценки длительности заболевания до перехода в острый лейкоз и общей продолжительности жизни больных результат цитогенетического анализа был расценен как важнейший прогностический признак. В группу с благоприятным прогнозом отнесены случаи с единичными хромосомными аномалия ми: -Y, 5q- и 20q-. Неблагоприятное течение наблюдали при множественных (сложных) нарушениях (три или более перестройки кариотипа) и изменениях хромосомы 7 (делеции длинного плеча, моносомии).
Другие аномалии определяли «промежуточный» прогноз. Средняя длительность заболевания до перехода в острый лейкоз составила по группам 9,4; 0,4 и 1,1—3,3 года соответственно. Эти данные используются при оценке эффективности новых схем лечения миелодисплазии и зарекомендовали себя как одна из лучших прогностических систем при миелодиспластическом синдроме.
Важное диагностическое значение может иметь метод FISH в тех случаях, когда стандартное цитогенетическое исследование неинформативно или обнаруживаются только единичные клетки с нарушением кариотипа, которое по формальным критериям нельзя считать клоном. Для диагностики наиболее характерных хромосомных нарушений при миелодиспластическом синдроме разрабатывается панель FISH-зондов.
Попытки выделить цитогенетические особенности каждой из клинико-морфологических субъединиц, входящих в общую гетерогенную группу миелодиспластических синдромов, не увенчались успехом. В то же время ХММЛ, рассматриваемый как миелопролиферативное заболевание с морфологическими признаками миелодисплазии, нередко ассоциируется со специфической хромосомной аномалией t(5;12)(q33;p13), однако в большинстве случаев ХММЛ эта хромосомная аномалия не выявляется.
— Читать далее «Хромосомные аномалии при хроническом миелолейкозе (ХМЛ) — прогноз»
Оглавление темы «Цитогенетика лейкозов»:
- Транслокация (8;14)(q24;q32) при остром лимфобластном лейкозе
- Транслокация (1;19)(q23;p13) при остром лимфобластном лейкозе
- Транслокация (12;21)(p13;q22) при остром лимфобластном лейкозе
- Делеция 6q (6q-), 9р (9р-) при остром лимфобластном лейкозе
- Прогностическое значение хромосомных аномалий при остром лимфобластном лейкозе
- Хромосомные аномалии при миелодиспластическом синдроме — прогноз
- Хромосомные аномалии при хроническом миелолейкозе (ХМЛ) — прогноз
- Цитогенетика бластного криза хронического миелоидного лейкоза — изменения кариотипа
- Дифференциальная диагностика хронического миелолейкоза
- Молекулярно-генетические методы оценки эффективности лечения хронического миелолейкоза
Источник
7-я хромосо́ма челове́ка — одна из 23 человеческих хромосом. Хромосома содержит более 158 млн пар оснований[1], что составляет от 5 до 5,5 % всего материала ДНК человеческой клетки. В настоящее время считается, что на 7-й хромосоме находятся от 1000 до 1400 генов.
7-я хромосома содержит кластер A генов гомеобокса.
Гены[править | править код]
Эта статья или раздел содержит незавершённый перевод с английского языка. Вы можете помочь проекту, закончив перевод. |
Ниже перечислены некоторые гены, расположенные на 7-й хромосоме:
Плечо p[править | править код]
- ABCA13 — 13-й член A-подсемейства АТФ-связывающих кассетных белков;
- AHR — рецептор ароматических углеводородов
- AQP1 — Аквапорин 1;
- C1GALT1 — гликозилтрансфераза;
- CBX3 — хромобокс гомолог 3;
- CCM2 — церебральная кавернозная мальформация 2;
- DFNA5 — глухота, аутосомно-доминантный тип 5;
- GARS — глицил-тРНК-синтетаза;
- IL6 — интерлейкин 6;
- ITGB8 — гликопротеин из надсемейства интегринов (β8);
- NOD1 — Nod-подобный рецептор подсемейства NOD;
- PMS2 — PMS2 англ. postmeiotic segregation increased 2 (S. cerevisiae).
Плечо q[править | править код]
- ABCB1 — P-гликопротеин;
- ASL — аргининосукцинат-лиаза;
- CAV1 — кавеолин 1;
- CCL24 — Chemokine (C-C motif) ligand 24 (scya24);
- CCL26 — Chemokine (C-C motif) ligand 26 (scya26);
- CD36;
- CDK5 — циклин-зависимая киназа 5;
- CGRP-RCP — calcitonin gene-related peptide receptor component protein;
- CFTR — Трасмембранный регулятор муковисцидоза, ATP-binding cassette (sub-family C, member 7);
- CLCN1 — хлоридный канал 1;
- CNTNAP2 — ген, ассоциированный с аутизмом;
- COL1A2 — коллаген, тип I, альфа 2;
- CYLN2 — cytoplasmic linker 2;
- DLD — дигидролипоамидная дегидрогеназа (E3 компонент пируват дегидрогеназного комплекса, 2-oxo-glutarate complex, branched chain keto acid dehydrogenase complex);
- ELN — эластин (надклапанный аортальный стеноз, Williams-Beuren syndrome);
- FOXP2 — Forkhead box protein 2;
- GTF2I — general transcription factor II, i;
- GTF2IRD1 — GTF2I repeat domain containing 1;
- GUSB — бета-глюкуронидаза;
- HSPB1 — heat shock 27kDa protein 1;
- KCNH2 — potassium voltage-gated channel, subfamily H (eag-related), member 2;
- KRIT1 — KRIT1, ankyrin repeat containing;
- LIMK1 — LIM domain kinase 1;
- NAMPT — никотинамидфосфорибозилтрансфераза;
- NOS3 — эндотелиальная синтаза оксида азота
- p47 phox или NCF1 — 47 kDa нейтрофил оксидазный фактор / нейтрофил цитозольный фактор 1;
- PIK3CG — каталитическая субъединица γ фосфатидилинозитол-4,5-бисфосфат-3-киназы (PI3K gamma, p110γ)[2];
- RELN — рилин;
- SBDS — Shwachman-Bodian-Diamond syndrome;
- SH2B2 — адаптерный белок;
- SLC25A13 — solute carrier family 25, member 13 (citrin);
- SLC26A4 — solute carrier family 26, member 4;
- SRI — sorcin;
- TAS2R16 — taste receptor, type 2, member 16;
- TFR2 — рецептор трансферрина 2;
- TPST1 — тирозилпротеин сульфотрансфераза 1;
- VGF — «индуцируемый фактором роста нервов».
Болезни и расстройства[править | править код]
Ниже перечислены некоторые заболевания, связанные с генами 7-й хромосомы, а также гены, дефекты которых вызывают эти заболевания:
- аргининосукциновая ацидурия (англ. argininosuccinic aciduria) — ASL;
- бессиндромная глухота (англ. nonsyndromic deafness) аутосомно-доминантный тип 5 и аутосомно-рецессивный тип 4 — DFNA5 и SLC26A4;
- болезнь мочи с запахом кленового сиропа — DLD;
- болезнь Шарко — Мари — Тута типов 2В и 2F — GARS и HSPB1;
- врожденное двустороннее отсутствие семявыносящих протоков (англ. congenital bilateral absence of vas deferens) — CFTR;
- гемохроматоз типа 3 (англ. hemochromatosis, type 3) — TFR2;
- дистальная спинальная амиотрофия (англ. distal spinal muscular atrophy) типа 5 — GARS;
- кавернозная ангиома (англ. cerebral cavernous malformation) — CCM2;
- миелодиспластический синдром;
- муковисцидоз — CFTR;
- мукополисахаридоз типа VII, или синдром Слая — GUSB;
- наследственный неполипозный колоректальный рак (англ. hereditary nonpolyposis colorectal cancer) — PMS2;
- несовершенный остеогенез типов I, II, III и IV — COL1A2;
- сахарный диабет взрослого типа у молодых типа 2 — GCK;
- синдром Вильямса — ASL, BAZ1B, BCL7B, CLDN3, CLDN4, CLIP2, EIF4H, ELN, FZD9, FKBP6, GTF2I, GTF2IRD1, HIP1, KCTD7, LAT2, LIMK1, MDH2, NCF1, NSUN5, POR, RFC2, STX1A, TBL2, TRIM50, TRIM73, TRIM74, WBSCR14, WBSCR18, WBSCR21, WBSCR22, WBSCR23, WBSCR24, WBSCR27 и WBSCR28;
- синдром Норман — Робертс — RELN;
- синдром Пендреда — SLC26A4;
- синдром Романо — Уорда (англ. Romano–Ward syndrome) — KCNH2;
- синдром Швахмана — Даймонда (англ. Shwachman–Diamond syndrome) — SBDS;
- синдром Элерса — Данлоса с артрохалазией типа 7B — COL1A2;
- хроническая гранулематозная болезнь, обусловленная недостаточностью нейтрофильного цитозольного фактора 1 — NCF1
- цитруллинемия (англ. citrullinemia) типа II — SLC25A13;
- шизофрения — KCNH2, ABCA13
Хромосомные болезни[править | править код]
Некоторые расстройства вызываются изменениями в структуре или количестве копий 7-й хромосомы:
- синдром Вильямса — делеция участка длинного плеча хромосомы в позиции 7q11.23, который содержит более 20 генов; потеря некоторых из этих генов и связана с характерными особенностями расстройства, однако для большинства генов удалённого участка связь с симптомами пока не установлена;
- задержка роста и развития, умственная отсталость, характерные изменения черт лица, скелетные аномалии, замедленная речь и другие медицинские проблемы — дополнительная копия части хромосомы (частичная трисомия) или отсутствие части хромосомы (частичная моносомия), иногда делеция или дупликация части хромосомы, а также возникновение кольцевой хромосомы (англ. ring chromosome).
Примечания[править | править код]
Источник
banner1
Моносомия по 7 хромосоме
Найдено (34 сообщения)
генетик
16 апреля 2014 г. / Наталия / Дрогобыч
… , Енштейн-Бар Вирусом, первичный Миело Диспластический Синдром, Моносомия 7.
Сейчас и гемоглобин и тромбоцыты в норме и не падают в течении … 14-04-2014 artlat, обратиться
Наталия,
люди с моносомией по 7 хромосоме не жизнеспособны. Тут какая то ошибка.
… открыть
16 апреля 2014 г. / Елена
Наталия, моносомия по 7-ой хромосоме имеет выявленная у Вашего ребёнка, имеет плохой прогноз в смысле перехода заболевания в острый лейкоз. То, что у ребёнка наблюдается видимое здоровье не снимает риска. Вы должны обратиться к хорошему … смотреть
генетик
10 ноября 2013 г. / Дарья / волгоград
… на скрининг узи увидели отек шеи плода. Беременность по мед. Показаниям прервали.
Результат гистологии: недостаточное количество митозов. Полная или частичная моносомия по 13 хромосоме (хорион 2). Полирлоидный набор хромосом (хорион … открыть
генетик
5 июля 2013 г. / Аноним / г. Братск
… D21S341)*2, при анализе обнаружена одна копия хромосомы Х и по две копии хромосомов 13 и 21. Т.е у плода обнаружена моносомия по хромосоме Х. Сказал врач, что надо пройти генетическое обследование пары. объясните пожалуйста диагноз и … открыть
генетик
7 мая 2013 г. / Светлана… / Санкт-Петербург
Добрый день ! Мне 27 лет .Мужу 33 года .Была замерзшая беременность на сроке 9 недель. Диагноз генетическая патология. моносомия по 21 хромосе Х.Какой шанс родить здорового… открыть
5 июля 2013 г. / Аноним
… D21S341)*2, при анализе обнаружена одна копия хромосомы Х и по две копии хромосомов 13 и 21. Т.е у плода обнаружена моносомия по хромосоме Х. Сказал врач, что надо пройти генетическое обследование пары. объясните пожалуйста диагноз и … смотреть
генетик
5 апреля 2013 г. / Наталья А.… / Самара
Здравствуйте! Мне 34 года, супругу 38 лет. В анамнезе 3 (самопроизвольных) замерших беременности на ранних сроках (6-7 недель, 5 недель и 8нед. и 5 дней- в послед. случае делали… открыть (еще 2 сообщения)
Последние 5:
7 апреля 2013 г. / Наталья А.…
… при норме 286… И только после этого по рекомендации репродуктолога я срочно связалась с гематологом, … любых хромосомных сбоев, причем необязательно по хромосомам 13 и 14? Скажем, почему у … 18 хромосоме, 1- по 21, 2 трисомии по 13 и 14 и 1 моносомия по 14- … смотреть
генетик
4 марта 2013 г. / Светлана… / СПб
Добрый день!
У меня сбалансированная инверсия по 3 хромосоме 46 хх ,inv (3) (g 13,2 … ХХ, не обнаружен дисбаланс аутосомы 3, моносомия 15, ост. иссл. аутосомы представлены … трактовать заключение » не обнаружен дисбаланс по аутосоме 3″ — как то, что кариотип … открыть (еще 1 сообщение)
Последние 5:
генетик
10 июля 2012 г. / Людмила / СПб
Здравствуйте. Мне 31 год.Замершая беременность 7-8 недель. Цитогенетическое исследование плода -заключение:моносомия Х хромосомы.(кариотип 45 х).Подскажите,пожалуйста,… открыть
10 июля 2012 г. / Елена
Людмила, скорее всего произошёл случайный сбой. Подобные сбои — моносомия по Х хромосоме — 45,ХО — синдром Тернера — являются наиболее частыми причинами ранних выкидышей.
Планировать следующую беременность возможно не ранее … смотреть
генетик
5 июля 2012 г. / Людмила / СПб
Здравствуйте. Замершая беременность 7-8 недель. Через сколько можно планировать следующую беременность? Цитогенетическое исследование плода -заключение:моносомия… открыть
6 июля 2012 г. / Елена
…
Наиболее распространённой причиной самопроизвольных выкидышей является моносомия по Х хромосоме, так называемый синдром Тернера — кариотип 46 … до 4-х мес. беременности.
Вести беременность нужно по стандартному протоколу. Однако, не взирая на результаты … смотреть
генетик
9 июня 2012 г. / Ольга / Санкт-Петербург
Добрый день. Подскажите пожалуйста. Мне 37 лет и 2 зам. беременности. 7-8 нед. .Проведена генетическое исследования интерфазных клеток ворсинок хориона. выявлена генет.… открыть
9 июня 2012 г. / Елена
Ольга, моносомия по 21 хромосоме, как собственно и любая другая моносомия, кроме моносомии по Х хромосоме, ведёт к неразвивающейся беременности и гибели плода на ранних стадиях его развития.
Вам и Вашему мужу … смотреть
Страницы: < 1 2 3 4 >
Портал зарубежной недвижимости JJC.Ru
Источник
Моносомия относится к генетическим аномалиям, для которых характерно изменение кариотипа. В норме у человека определяется 23 хромосомы, каждая из которых имеет гомологичную пару. Если одна из них лишается своей пары, то развивается моносомия. Заболевание протекает тяжело, часто приводит к внутриутробной гибели плода. В других случаях ребенок рождается живым, но при этом наследует тяжелые врожденные изменения, которые объединяют в синдромы, самыми распространенными из которых являются синдром Шерешевского-Тернера и кошачьего крика.
Синдром Шерешевского-Тернера
Является следствием моносомии по хромосоме Х. Больные дети часто рождаются недоношенными или имеют сниженную массу тела. Одним из классических признаков синдрома Шерешевского-Тернера, который можно заметить сразу после рождения, является выраженная кожная складка на шее. Среди других клинических проявлений отмечаются:
- пороки сердца;
- отечность верхних и нижних конечностей;
- нарушение циркуляции лимфы;
- задержка речевого и физического развития.
По мере взросления ребенка проявляются характерные черты строения тела. Рост обычно не превышает 150 см, крыловидные складки на шее сохраняются, ушные раковины могут деформироваться, верхняя челюсть недоразвита, грудная клетка широкая. Моносомия по хромосоме Х влияет на развитие органов половой системы. У женщин отмечается отсутствие фолликулов в яичниках, нарушение менструального цикла, недоразвитие молочных желез. У мужчин снижается уровень тестостерона, может отсутствовать одно или оба яичка либо отмечаться их недоразвитие.
Прогноз при синдроме Шерешевского-Тернера относительно благоприятный. При отсутствии тяжелых пороков развития и регулярном наблюдении у специалиста продолжительность жизни не сокращается.
Синдром кошачьего крика
Является примером частичной моносомии. В данном случае теряется не вся хромосома из одной пары, а только определенный участок — короткое плечо 5-й хромосомы. Заболевание получило свое название из-за специфического плача, который издают новорожденные дети. Он обусловлен недоразвитием гортани и ее хрящевых компонентов. Кроме того, выявляются и другие симптомы данного вида моносомии:
- отклонение в умственном и физическом развитии;
- изменение формы головы и черт лица (недоразвитая нижняя челюсть, специфический внешний вид глаз и ушных раковин);
- недостаток массы тела;
- врожденные пороки развития (микроцефалия, пороки сердца, мышц, внутренних органов).
Как и предыдущий вид моносомии, синдром кошачьего крика характеризуется благоприятным течением при условии, что отсутствуют тяжелые пороки, которые могут стать причиной летального исхода.
Причины моносомии
Моносомия может возникать на различных стадиях клеточного деления. Например, при синдроме Шерешевского-Тернера нарушается процесс расхождения Х-хромосом. В результате в одну яйцеклетку женщины попадает две Х-хромосомы, а во вторую ни одной. Во время процесса оплодотворения зигота получает набор Х0 и Y0,- вместо нормального ХХ или XY.
Причины появления моносомии не связаны с наследственными факторами. Нарушения возникают при воздействии неблагоприятных факторов. Оказывать влияние на половые клетки могут вредные привычки, радиация, некоторые лекарственные препараты, химические вещества, неблагоприятная экологическая обстановка, вредные условия труда и т. д.
Диагностика моносомии
Выявить заболевание можно еще на этапе внутриутробного развития. Для этого всем беременным женщинам проводится скрининговое УЗИ. Если специалист выявляет нарушения развития плода, то дополнительно назначается биопсия хориона, которая позволяет получить образец ткани и определить кариотип. Таким способом можно выявить не только моносомии, но и другие генетические нарушения.
Сделать кариотипирование вы можете в медико-генетическом центре «Геномед». Здесь современное точное оборудование и опытные специалисты. Такое сочетание позволяет получить достоверные результаты, необходимые для постановки диагноза.
Источник