Синдром мириззи код мкб

Синдром Мириззи — редкое осложнение желчнокаменной болезни, характеризующееся частичным сужением общего печеночного или желчного протока в результате воспаления в нём и сдавления извне желчным конкрементом, расположенным в пузырном протоке или шейке желчного пузыря. Всё это в дальнейшем может привести к развитию стриктуры гепатикохоледоха или к образованию фистулы между желчным пузырём и гепатикохоледохом[1].
Классификация[править | править код]
В настоящее время используют классификацию в 1989 г. по степени разрушенности стенки общего печеночного протока пузырнохоледохеальным свищем:
I тип – компрессия общего печеночного протока камнем шейки желчного пузыря или пузырного протока;
II тип – пузырно-холедохеальный свищ, занимающий менее 1/3 окружности общего печеночного протока;
III тип – пузырно-холедохеальный свищ, занимающий 2/3 окружности общего печеночного протока;
IV тип – пузырно-холедохеальный свищ, занимающий всю окружность общего печеночного протока (стенка протока полностью разрушена).
Этиология[править | править код]
Точная причина заболевания не известна, однако, основной причиной развития болезни принято считать хронический калькулезный холецистит, при котором происходит миграция конкремента в карман Хартмана, шейку пузыря или пузырный проток, плотно прилегая к стенке и формируя пролежень. Не мало важными факторами являются близкие анатомические расположения желчного пузыря, пузырного протока и гепатохоледоха. При этом вероятность возникновения болезни возрастает при параллельном расположении пузырного и печеночного протоков, при наличии единой оболочки, покрывающей оба канала. Некоторые авторы выделяют врожденную аномалию развития желчевыводящей системы, при которой пузырный проток имеет большую длину и впадает в нижнюю треть гепатохоледоха. В этом случае, при наличии конкремента в пузырном канале, увеличивается риск сдавления общего протока печени и развития патологического синдрома[2].
Клиническая картина[править | править код]
Для синдрома Мириззи в основном характерны симптомы острого и хронического холецистита с развитием механической желтухи. В анамнезе больных отмечается длительное течение желчнокаменной болезни с частыми приступами, перемежающимися периодами механической желтухи.
Выделяют 3 основных симптома синдрома Мириззи:
- Боль в верхнем правом квадранте живота;
- Желтуха;
- Симптомы холангита (озноб, повышение температуры тела)[3].
Диагностика[править | править код]
- Ультразвуковое исследование (диагностическая ценность 8-67%);
- МРТ (диагностическая ценность 94,4%);
- Эндоскопическая ретроградная холангиопанкреатография;
- Чрескожно-чреспеченочная холангиография.
Лечение[править | править код]
Основное лечение синдрома Мириззи — хирургическое.
- при I типе – холецистэктомия, ревизия желчных протоков с дренированием Т-образной трубкой;
- при II, III типах – холецистэктомия, удаление конкрементов, ушивание свищевого отверстия атравматическим рассасывающимся шовным материалом или пластика дефекта оставленной частью культи желчного пузыря, дренирование Т-образной трубкой;
- при IV типе– предпочтительнее гепатикоеюностомия с петлёй тощей кишки по Ру, так как стенка общего печеночного протока полностью разрушена.
Примечания[править | править код]
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Крузона.
Синдром Крузона
Описание
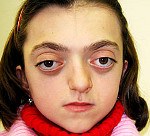
Синдром Крузона. Редкое генетическое заболевание, сопровождающееся прогрессирующими деформациями лицевой и мозговой части черепа и краниосиностозом с развитием сопутствующих нарушений. Симптомами этого состояния являются изменение формы головы (брахицефалия, скафоцефалия, тригоноцефалия), крючковидный нос, гипоплазия средней трети лица, нарушения зрения и слуха. Диагностика синдрома Крузона осуществляется на основании внешних проявлений заболевания, рентгенологических данных, а также молекулярно-генетических анализов. Специфического лечения этой патологии не существует, используются паллиативные и симптоматические мероприятия, в том числе хирургического характера.
Дополнительные факты
Синдром Крузона (краниофасциальный дизостоз 1 типа) – генетическое заболевание, характеризующееся нарушением процессов окостенения и развития элементов скелета лицевого и мозгового черепа. Впервые это состояние было описано в 1912 году французским педиатром О. Крузоном, с тех пор синдром носит его имя. Механизм наследования синдрома Крузона – аутосомно-доминантный, однако заболевание часто обусловлено спонтанными мутациями. Патология встречается достаточно редко – примерно 1,6 случаев на 100 000 новорожденных, при этом данным синдромом обусловлено почти 5% от всех пороков развития, сопровождающихся черепным дизостозом. Долгое время считалось, что это состояние имеет две разновидности – обычную и сопровождающуюся кожными нарушениями (гиперкератозом, акантозом), но с учетом современных данных специалисты в области генетики установили, что синдром Крузона с черным акантозом (CAN) является отдельным наследственным заболеванием. В то же время, патогенез его развития аналогичен классической форме заболевания, именно этим объясняется значительная схожесть симптомов. Состояние с одинаковой вероятностью поражает как мальчиков, так и девочек.
Синдром Крузона
Причины
Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме – он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов, что снижает его стабильность – в нем часто развиваются дефекты, приводящие к многочисленным генетическим заболеваниям, в основном поражающим элементы скелета. Так, помимо синдрома Крузона, мутации FGFR2 могут быть причиной синдромов Апера, Сетре-Чотзена, Бира-Стивенсона, синдрома Пфайффера и многих других патологий. Генетические исследования показали, что краниофасциальный дизостоз 1-го типа способны вызывать более 35 мутаций вышеуказанного гена, в основном они локализованы в области 7 и 9 экзонов.
Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка. Изменение конформации рецептора к фактору роста фибробластов 2 нарушает межклеточные взаимодействия в соединительных тканях черепа, главным образом костной и хрящевой. Это приводит сначала к накоплению фибробластов в области межкостных швов, а потом к активизации процессов окостенения, что и является причиной ведущего проявления синдрома Крузона – черепного синостоза. Некоторые исследователи полагают, что данные генетические дефекты влияют также на эмбриональное развитие структур первой жаберной дуги – к ним относят челюсти и отчасти элементы средней трети лица. Именно этим объясняется гипоплазия челюстей, особенно нижней, при синдроме Крузона.
Причины синдрома Крузона с черным акантозом несколько иные – он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями – Ala391Glu Это тоже миссенс-мутация, изменяющая структуру белка-рецептора. Патогенез заболевания практически не отличается от классического варианта. Изменения лица и черепа при синдроме Крузона с черным акантозом аналогичны предыдущему типу, однако к ним присоединяются гиперкератоз различных участков кожи и акантоз, нередко наблюдаются многочисленные родинки.
Симптомы
Проявления синдрома Крузона можно заметить уже при рождении ребенка, однако наиболее выраженными они становятся на протяжении первых 3-4 лет жизни. Самым характерным симптомом заболевания является краниосиностоз, который может развиваться на венечном или стреловидном (намного реже) шве, прочно соединяя кости и останавливая нормальный рост головы. Сразу после рождения первые признаки синостоза могут быть стертыми, но всегда наблюдается гипертелоризм, прогнатия нижней челюсти, изменение формы носа по типу «клюва попугая», незначительный экзофтальм из-за уменьшенного размера глазниц, низкое расположение наружного слухового прохода. Иногда при синдроме Крузона выявляется синдактилия пальцев, в этом случае необходимо производить дифференциальную диагностику с синдромом Апера. У некоторых больных обнаруживается атрезия хоан, затрудняющая дыхание, а также гидроцефалия, еще больше осложняющая течение заболевания за счет резкого возрастания внутричерепного давления.
Особенностью синдрома Крузона является неминуемое прогрессирование заболевания, особенно в отношении формы черепа. Из-за образования прочного синостоза и продолжающегося роста размеров головного мозга форма головы изменяется, возникает брахицефалия или «башенный череп» – в зависимости от того, по какому шву произошло срастание. При синдроме Крузона в области сросшихся костей черепа также могут образовываться экзостозы. Такая деформация приводит и к поражению органов зрения – сначала возникает расходящееся косоглазие, затем экзофтальм сильно прогрессирует вплоть до выпадения глазных яблок из орбиты. Нередко синдром Крузона сопровождается расстройствами слуха из-за нарушения структуры пирамиды височной кости – ее полости уменьшены в размерах, некоторые из них могут отсутствовать, нередко это приводит к полной глухоте. Наблюдаются изменения и со стороны нервной системы, обнаруживаются нарастающие признаки умственной отсталости (при отсутствии паллиативных мероприятий), симптомы повышения внутричерепного давления (головные боли, рвота), судорожные припадки.
Рвота. Судороги.
Диагностика
Выявление синдрома Крузона возможно на этапе пренатального развития, сразу после рождения или в первые годы жизни больного. Для этого применяются рентгенологические методики, общий осмотр, молекулярно-генетические анализы. Вспомогательную роль в диагностике синдрома Крузона играют такие методы, как офтальмологический осмотр, исследование слуха, оценка интеллектуального и психического развития. При осмотре маленьких детей определяются низко посаженные уши, гипоплазия средней трети лица, экзофтальм. У больных синдромом Крузона старшего возраста к этим проявлениям присоединяются расходящееся косоглазие, ослабление слуха вплоть до полной глухоты, изменение формы черепа. На рентгенографии черепа регистрируется синостоз в области венечного, стреловидного или лямбдовидного швов, возможно обнаружение экзостозов и уплощенной формы глазниц.
Томография пирамиды височной кости при синдроме Крузона выявляет нарушение формирования наружного слухового прохода (атрезия или стеноз) и других полостей, иногда наблюдается отсутствие барабанной полости. Турецкое седло несколько расширено, могут образовываться добавочные мелкие околоносовые синусы. Молекулярно-генетическая диагностика синдрома Крузона производится врачом-генетиком и при классической форме заболевания сводится к автоматическому секвенированию 7 и 9 экзонов гена FGFR2 с целью выявления мутаций. При наличии кожных проявлений (гиперкератоза, бородавках, множественных родинках) имеет смысл производить поиск мутации Ala391Glu в гене FGFR3. Для обеих форм синдрома Крузона возможна пренатальная генетическая диагностика, ультразвуковые методики при этом, как правило, малоэффективны.
Лечение
Какого-либо специфического лечения синдрома Крузона на сегодняшний момент не существует, применяют только паллиативные мероприятия. К ним относят хирургические вмешательства по ремоделированию формы черепа и устранению синостозов – такие процедуры необходимо начинать как можно раньше и в дальнейшем производить еще несколько раз по мере роста головы. Это снижает уровень внутричерепного давления, что положительно сказывается на умственном развитии больных синдромом Крузона и уменьшает вероятность появления неврологических нарушений. Также с помощью хирургических методик создают искусственный блефарофимоз для снижения степени экзофтальма и предотвращения вывиха глазного яблока. При атрезии хоан производится их расширение оперативным путем для облегчения дыхания. Описаны техники радикальных комплексных операций, направленных на устранение большинства лицевых нарушений при синдроме Крузона. В случае развития кожных изменений для снижения их выраженности рекомендуется наружное применение средств на основе ретиноидов, иногда назначают кортикостероиды.
Прогноз синдрома Крузона, как правило, неопределенный, многие специалисты оценивают его как неблагоприятный. Это связано с тем, что даже при проведении всех симптоматических и паллиативных мероприятий у больных все равно нарастает расходящееся косоглазие, практически всегда со временем развивается глухота, гипоплазия средней трети лица становится более выраженной с возрастом. Тем не менее, многие больные при соответствующем лечении и уходе могут доживать до преклонного возраста. По причине сильного нарушения зрения и слуха практически всегда происходит инвалидизация пациентов, причиной инвалидности также может стать умственная отсталость. Профилактика синдрома Крузона не разработана, возможно лишь пренатальное определение патологии молекулярно-генетическими методами.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Синдром Мириззи в это редкое осложнение , в котором желчный становится разорвались в пузырного протока или шейки желчного пузыря вызывает сжатие общего желчного протока (CBD) или общего печеночного протока , что приводит к непроходимости и желтухи. Желтуха может быть вызвана прямой внешней компрессией с помощью камня или из фиброза , вызванный хроническим холециститом (воспаление). Cholecystocholedochal свищ может произойти.
презентация
Синдром Мириззи не имеет ни одного согласующихся или уникальные клинические особенностей , которые отличают его от других более распространенных форм желтухи. Симптомы рецидивирующей холангит, желтуха, боли в правом верхнем квадранте, а также повышенной билирубина и щелочной фосфатазы может быть или может не присутствовать. Острые презентации синдрома включают симптомы , согласующиеся с холециститом .
Хирургия чрезвычайно трудно, как треугольник Кало в часто полностью уничтожены и риски причинения вреда в КБР высоки.
патофизиология
Несколько и крупные камни в желчный пузырь может стать разорвались в сумке Гартмана из желчного пузыря , что приводит к хроническому воспалению — что приводит к сжатию общих желчного протока (CBD), некрозу , фиброзу, и в конечном счете свищ образования в соседний общий печеночный проток (ИБС) или общий желчный проток (CBD). В результате, КБС / CBD становится затруднен либо рубцом или камнем, в результате чего обструктивной желтухи . Его можно разделить на четыре типа.
нет Тип I — Нет свищей подарок
- Тип IA — Наличие пузырного протока
- Тип IB — облитерации пузырного протока
Типы II-IV — Свищ настоящее
- Тип II — дефекты меньше, чем 33% от диаметра CHD
- Тип III — Дефект 33-66% от диаметра CHD
- Тип IV — дефект больше, чем 66% от диаметра CHD
диагностика
Изображения с помощью УЗИ , MRCP или КТ обычно делают диагноз. MRCP могут быть использованы для определения поражения анатомический до операции. Иногда синдром Мириззи диагностируется или подтвержден ЭРХПГОМ при запросе , чтобы облегчить обструктивную желтуху или холангит с помощью эндоскопа помещенного стент, или когда USS было ошибочно сообщаются как choledocolithiasis.
лечение
Простая холецистэктомия подходит для пациентов типа I. Для типов II-IV, субтотальная холецистэктомия может быть выполнена, чтобы избежать повреждений основных желчных протоков. Холецистэктомия и bilioenteric анастомоз может потребоваться. Ру-ан-Y гепатикоеюностомию показал хорошие результаты в некоторых исследованиях.
эпидемиология
Синдром Мириззите в происходит приблизительно в 0,1% пациентах с желчными камнями . Он находится в 0,7 до 2,5 процентов от холецистэктомии .
Это влияет на мужчин и женщин в равной степени, но, как правило, влияет на пожилых людей чаще. Там нет никаких доказательств того, рас, имеющих какое-либо отношение к эпидемиологии.
эпоним
Она названа в честь Пабло Луис Мириззи (1893-1964), аргентинского врача. Мириззи получил образование и обучения в своем родном городе, а затем посетил некоторые из лучших больниц по всей территории Соединенных Штатов для дальнейшего образования и профессиональной подготовки. Мириззи специализируется на брюшной и грудной хирургии и будет писать многоплодно по связанным с хирургическими темам.
Рекомендации
внешняя ссылка
- Мириззи синдром Разъяснения в USMLE форуме
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Ретта.
Синдром Ретта
Описание
Синдром Ретта. Генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника. Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного.
Дополнительные факты
Синдром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году. Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера.
Синдром Ретта
Причины
Этиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается. Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани.
При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей. Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК. В результате этого у больных синдромом Ретта наблюдается значительное превалирование процессов торможения в головном мозге, что отражается не только на физиологии центральной нервной системы, но и на ее морфологическом строении.
Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко. Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца. Дефекты этого гена приводят и другим патологиям центральной нервной системы – варианту Запела, синдрому Луба (Х-сцепленная умственная отсталость у мальчиков), врожденной энцефалопатии. Некоторые исследователи относят эти состояния к атипичным формам синдрома Ретта.
Симптомы
У новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям. Врачи-педиатры отмечают более медленный, нежели в норме, рост стоп и кистей в длину и замедление роста окружности головы. Иногда помимо неврологических проявлений может отмечаться нарушение работы печени, сердца, желудочно-кишечного тракта.
Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе. Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения.
Судороги. Эхолалия.
Диагностика
Диагностика синдрома Ретта производится на основании изучения анамнеза больного, его настоящего статуса, магнитно-резонансной томографии и энцефалографии, молекулярно-генетических анализов. Изучение наследственного анамнеза, как правило, не имеет особого смысла по причине спорадического характера мутаций гена MECP2. Характерными для синдрома Ретта являются нормальное развитие ребенка до 6-12 месяцев, возникновение мышечной гипотонии и беспокойства в раннем детстве, появление в дальнейшем атаксии и частых эпилептических припадков, стремительное утрачивание приобретенных навыков. В дальнейшем у больных регистрируется тяжелая умственная отсталость, мышечная слабость (вплоть до атрофии), искривление позвоночника, судорожные припадки.
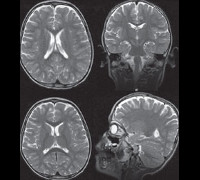
При осмотре больных синдромом Ретта выявляется отставание в росте и его остановка, резкое уменьшение окружности головы, отсутствие речи (на начальных этапах патологии характерна эхолалия). На магнитно-резонансной томографии головного мозга обнаруживается уменьшение размера органа, нечеткая дифференциация серого и белого вещества, базальных ганглиев, снижение складчатости коры больших полушарий. Электроэнцефалограмма подтверждает снижение фоновой активности головного мозга и резко ослабленную реакцию на внешние раздражители. Наиболее точную диагностическую информацию дают методы современной генетики – поиск делеций в локусе гена MECP2 или прямое секвенирование его последовательности для определения мутаций. Такое подтверждение синдрома Ретта возможно и в рамках пренатальной диагностики генетических заболеваний. Вспомогательную роль в установлении этого состояния может играть обследование внутренних органов (например, методами УЗИ) – у 20-30% больных выявляется недоразвитие печени или селезенки.
Лечение
Специфического лечения синдрома Ретта на сегодняшний день не существует. Имеются обнадеживающие данные некоторых исследовательских лабораторий, сотрудникам которых удалось «включить» ген MECP2 у мышей и тем самым добиться исчезновения симптомов заболевания. В сфере практической медицины пока доступна только симптоматическая терапия, однако и она сопряжена с рядом трудностей – в частности, эпилептические припадки при этом заболевании плохо поддаются устранению противосудорожными средствами. Также для лечения синдрома Ретта применяют ноотропные препараты, нарушения сна корректируют снотворными препаратами из группы барбитуратов или мелатонином.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник