Синдром миллера дикера по мкб 10

Рубрика МКБ-10: Q04.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q00-Q07 Врожденные аномалии пороки развития нервной системы / Q04 Другие врожденные аномалии (пороки развития) мозга
Определение и общие сведения[править]
Агенезия прозрачной перегородки
Редкая аномалия, встречают 2-3 случая на 10 000 населения.
Этиология и патогенез[править]
Причины отсутствия прозрачной перегородки
• Сочетание с другими мальформациями головного мозга, наиболее часто с агенезией мозолистого тела.
• Сочетание с аномалией зрительных трактов (септо-оптическая дисплазия).
• Выраженная гидроцефалия, когда происходит повреждение прозрачной перегородки (стеноз водопровода, аномалия АК II).
• Ишемически-гипоксическое поражение мозга.
Клинические проявления[править]
Другие редукционные деформации мозга: Диагностика[править]
МРТ
На срезах в корональной плоскости прозрачная перегородка не видна, вследствие чего желудочки сообщаются между собой, своды расположены ниже обычного, крыша боковых желудочков имеет горизонтальное положение. На аксиальных срезах отсутствует передняя вогнутость между лобными рогами боковых желудочков. На срединном сагиттальном изображении наиболее отчетливо видно низкое расположение сводов.
КТ
При КТ прозрачную перегородку не определяют, желудочки на уровне передних рогов сообщаются между собой.
УЗИ
Для диагностики агенезии прозрачной перегородки в пре- и постнатальном периоде необходимо использовать корональные срезы. Лобные рога боковых желудочков имеют квадратную или трапециевидную форму, сообщаются между собой. Своды расположены ниже обычного.
Алгоритм исследования
Агенезию прозрачной перегородки одинаково хорошо определяют при проведении пренатальной сонографии и МРТ. Преимущество пренатального МРТ — более точное определение положения сводов, для чего используют срезы в сагиттальной плоскости.
Дифференциальный диагноз[править]
Изолированную агенезию прозрачной перегородки необходимо дифференцировать с септо-оптической дисплазией. При септо-оптической дисплазии отсутствие прозрачной перегородки сочетается с гипоплазией зрительных трактов.
В случае обнаружения агенезии прозрачной перегородки необходимо исключить/подтвердить наличие других сочетанных аномалий мозга.
Другие редукционные деформации мозга: Лечение[править]
Профилактика[править]
Прочее[править]
Cиндром Жубера
Определение и общие сведения
Синдром Жубера характеризуется врожденными пороками развития мозгового ствола и недоразвитием или гипоплазией червя мозжечка, приводящих к аномальному дыханию, нистагму, гипотонией, атаксией и задержкой моторного развития.
Распространенность оценивается примерно в 1 / 100,000 новорожденных. Наследование атосомно-рецессивное.
Этиология и патогенез
Синдром генетически разнороден. Семь генов: AHI1 (6q23), NPHP1 (2q13), CEP290 (12q21), TMEM67 (8q22), RPGRIP1L (16q12), ARL13B (3p12.3-q12.3) и CC2D2A (4p15), а также два локуса: 9q34 ( JBTS1 ) и 11p12-Q13 ( CORS2 / JBTS2 ) — связывают с этим заболеванием на сегодняшний день.
Клинические проявления
В неонатальном периоде, болезнь часто проявляется нерегулярным дыханием (эпизоды тахипноэ и/или апноэ) и нистагмом. В младенчестве может развиваться гипотония, позже появляются симптомы мозжечковой атаксии (шаткая походка и дисбаланс). Задержка приобретения моторных навыков является распространенным проявлением синдрома жубера. Когнитивные способности варьируются, начиная от серьезного интеллектуального дефицита до сохранного интеллекта. Нейроофтальмологическое обследование может указать на наличие глазодвигательной апраксии. В некоторых случаях возникают судорожные припадки. Тщательное изучение лица демонстрирует характерный внешний вид: большая голова, выпуклый лоб, высокие округлые брови, веконосовые складки, птоз (иногда), нос задран вверх и имеет выпуклые ноздри, открытый рот (который, на ранней стадии имеет, как правило, овальную форму, позже приобретает «ромбовидный» внешний вид, и, наконец, потом становится треугольной формы с углами книзу), наблюдаются протрузия языка и ритмичные движения языка, иногда низко расположенные и наклоненные уши. Другие симптомы, которые иногда присутствуют при синдроме Жубера включают дистрофию сетчатки, нефронофтиз и полидактилию.
Диагностика
Диагноз основывается на обнаружении основных клинических признаков: гипотония, атаксия, задержка развития и глазодвигательная апраксия, которые должны сопровождаться наличием нейрорадиологического отличительного симптома — знака «коренного зуба» на магнитно-резонансной томографии, который является следствием гипоплазии червя мозжечка и пороков формирования среднего и заднего отделов мозга.
Дородовая диагностика возможна с помощью молекулярного анализа и УЗИ и МРТплода). Пренатальная диагностика показана семьям, в которых оба болезнетворные мутации были ранее идетифицированы.
Генетическое консультирование является важным клиническим инструментом предотвращения новых случаев, особенно для пар, у которых выявлено заболевание первого ребенка. Риск появления больного ребенка в дальнейшм составляет 25%.
Дифференциальный диагноз
Дифференциальный диагноз включает расстройства, связанные с синдромом Жубера, пороки развития червя мозжечка без знака «коренного зуба» (Денди-Уокера мальформация), Х-сцепленная гипоплазия мозжечка, атаксия с глазодвигательной апраксией типа 1 и 2, врожденные нарушения гликозилирования, 3C синдром, мостомозжечковые гипоплазии/атрофии, рото-лице-пальцевой синдром типа II и III, и синдром Меккеля-Грубера.
Лечение
Лечение является симптоматическим. Программы в области образования, физической, профессиональной и речевой терапии могут уменьшить гипотонию и сократить задержку в достижении моторных навыков.
Прогноз
Прогноз благоприятный для легких форм заболевания. Лечение больных с более тяжелыми формами следует проводить в специализированном центре.
Гипоплазия и агенез мозжечка
Определение и общие сведения
Гипоплазия мозжечка описывается в контексте различных заболеваний: хромосомные аномалии, внутриутробное воздействия токсинов и инфекционных агентов, нарушения обмена веществ и широкий спектр редких генетических и неврологических заболеваний. Гипоплазия мозжечка может затрагивать область червя и/или полушарий мозжечка и варьирует от частичного до полного недоразвития. Гипоплазия мозжечка может быть ограничена только областью мозжечка (гипоплазия гранулированных клеток типа Нормана, мальформация Данди-Уокера) или влиять на другие структуры ЦНС: средний мозг (синдромы «коренного зуба»), мост и продолговатый мозг (мосто-мозжечковая гипоплазия), кору головного мозга (лиссэнцефалия и гипоплазия мозжечка). Различие между гипоплазией и атрофией мозжечка не всегда очевидно, поскольку вторичная атрофия может сопутствовать гипоплазии мозжечка. Описаны также синдромы с гипоплазией мозжечка и пороками развития почек, глаз, печени или сердца: Гиллеспи, Ритшера-Шинцеля, рото-лице-пальцевой синдром типа II, Хойераала-Хрейдарссона.
Этиология и патогенез
Наследование может быть аутосомно-рецессивным, аутосомно-доминантным или Х-сцепленным. Генные мутации были идентифицированы при лиссэнцефалии и гипоплазии мозжечка (reelin), мосто-мозжечковой гипоплазии (PMM2), Х-сцепленной гипоплазии мозжечка (OPHN1, DCK1) и несколько локусов были нанесены на карту генома для аутосомно-рецессивных атаксии. Мутации фактора транскрипции поджелудочной железы (PTF1A) были идентифицированы в семье с агенезом поджелудочной железы и мозжечка. Гетерозиготные потери генов ZIC1 и ZIC 4 ответственны за развитие в мальформация Данди-Уокера.
Клинические проявления
Наиболее распространенные клинические проявления гипоплазии мозжечка: задержка развития речи, гипотония, атаксия и аномальные движения глаз.
Диагностика
Диагноз должен быть подтвержден визуализацией области мозжечка и головного мозга и длительным сроком клинического наблюдения.
Лечение и прогноз
Психический статус является важным элементом прогноза. В большинстве случаев никакого конкретного лечения не существует.
Синдром Уокера-Варбурга
Определение и общие сведения
Синдром Уокера-Варбурга — редкая форма врожденной мышечной дистрофии, сопровождаемой аномалиями развития мозга и глаз.
Распространенность синдрома Уокера-Варбурга оценивается в 1/60 500 человек. Наследование является аутосомно-рецессивным.
Этиология и патогенез
Синдром Уокера-Варбурга вызван аномальным O-гликозилированием альфа-дистрогликана, что приводит, помимо развития аномалий мозга, к врожденной мышечной дистрофии. Синдром Уокера-Варбурга представляет собой наиболее тяжелый фенотип так называемых дистрогликанопатий. Несколько генов вовлечены в этиологию синдрома Уокера-Варбурга. Большинство мутаций были обнаружены в генах белка O-маннозилтрансферазы 1 и 2 (POMT1 и POMT). Другие гены пути гликозилирования альфа-дистрогликана (FKTN, LARGE, FKRP и POMGNT1) выявляли мутации в случаях синдрома Уокера-Варбурга. Мутация в гене COL4A1, непосредственно не связанная с посттрансляционной модификацией дистрогликана, также была идентифицирована у пациентов с синдромом Уокера-Варбурга.
Клинические проявления
Пациенты проявляются при рождении тяжелой генерализованной гипотонией, мышечной слабостью, отсутствием или очень низким психомоторным развитием, судорогами и вовлечением глаз. МРТ головного мозга выявляет лиссэнцефалия по типу «булыжной мостовой» (2-й тип), гидроцефалию, тяжелую гипоплазию ствола мозга и мозжечка (возможна мальформация Дэнди-Уокера). Наблюдаются также аномалии белого вещества.
Диагностика
Лабораторные исследования обычно выявляют повышенную креатинкиназу, миопатическую/ дистрофическую патологию мускулатуры с измененной альфа-дистрогликановой экспрессией. Антенатальная диагностика возможна в семьях с известными мутациями.
Дифференциальный диагноз
Дифференциальный диагноз синдрома Уокера-Варбурга включает другие типы врожденных мышечных дистрофий и миопатий.
Лечение
Специального лечения синдрома Уокера-Варбурга нет. Лечение является только поддерживающим.
Прогноз
Синдром Уокера-Варбурга является самой тяжелой формой врожденной мышечной дистрофии, большинство детей умирает до достижения трехлетнего возраста.
Cиндром Миллера-Дикера
Определение и общие сведения
Cиндром Миллера-Дикера представляет собой синдром делеции хромосомы 17p13.3, характеризуется наличием классической лиссэнцефалии 1 типа и харктерными особенностями черт лица. Дополнительные врожденные пороки развития также могут быть частью состояния.
Cиндром Миллера-Дикера является редким заболеванием, расспространненость оценивается на уровне 1 случая на 100 000 новорожденных, хотя возможно она в реальности выше.
Этиология и патогенез
Видимые и субмикроскопические делеции 17p13.3, в том числе гена LIS1, встречаются почти у 100% пациентов.
Клинические проявления
Пациенты демонстрируют тяжелую задержку развития, как правило, страдают эпилепсией и недостаточным весом. Наблюдается лиссэнцефалия, либо генерализованная агирия или лобные агирия или пахигирия.
Лечение
Лечение только симптоматическое. Во избежание развития алиментарной недостаточности и аспирационной пневмонии — используют назогастральный зонд или гастростому. Терапия приступов эпилепсии имеет важное значение в ведении таких больных.
Нарушения миграции нейронов
Нарушения миграции нейронов представляют собой целую группу врожденных генетических дефектов, вызванных аномалиями миграции нейронов в развивающемся мозге и нервной системе.
Структурные аномалии, обнаруженные при нарушении миграции нейронов, включают в себя шизэнцефалию, порэнцефалию, лиссэнцефалию, агирию, макро- и микрогирию, полимикрогирию, пахигирию, нейронные гетеротопии, агенез мозолистого тела и черепных нервов.
Мутации многих генов участвуют развитии патологии, ген DCX при классической лиссэнцефалии, TUBA1A при микролиссэнцефалии с агенезом мозолистого тела, RELN и VLDLR при лиссэнцефалии с гипоплазией мозжечка. Мутации в гене ARX вызывают целый ряд фенотипов, начиная от гидроэнцефалии или лиссэнцефалии до эпилептических энцефалопатий с ранним началом , включая синдром Охтахара и инфантильные спазмы или умственную неполноценность без каких-либо пороков развития мозга.
Источники (ссылки)[править]
Лучевая диагностика и терапия заболеваний головы и шеи [Электронный ресурс] / Трофимова Т.Н. — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970425695.html
«Патологическая анатомия [Электронный ресурс] : национальное руководство / гл. ред. М.А. Пальцев, Л.В. Кактурский, О.В. Зайратьянц — М. : ГЭОТАР-Медиа, 2014. — (Серия «Национальные руководства»).» — https://www.rosmedlib.ru/book/ISBN9785970431542.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Содержание
- Описание
- Дополнительные факты
- Симптомы
- Диагностика
- Причины
- Профилактика
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Болезнь Келера.
Болезнь Келера
Описание
Болезнь Келера. Хроническое дистрофическое заболевание костей стопы, приводящее к их асептическому некрозу. Заболевание может протекать с поражением ладьевидной кости (болезнь Келера I) или плюсневых костей (болезнь Келера II). Болезнь Келера проявляется отечностью и болями в стопе в области пораженной кости, усилением болевого синдрома при ходьбе и его прогрессированием с течением времени, изменением походки и хромотой при одностороннем поражении. Характерным признаком является отсутствие воспалительных изменений пораженной области. Диагностика заболевания основывается на данных рентгенологического исследования. Лечение состоит в снятии нагрузки с пораженной стопы путем ее иммобилизации и последующей восстановительной терапии (лечебная гимнастика, физиотерапия, массаж).
Дополнительные факты
Заболевание, описанное Келером в 1908 году, получило название болезнь Келера I. Оно представляет собой асептический некроз ладьевидной кости, в то время как в травматологии известна также болезнь Келера II — асептический некроз головок костей плюсны. Дегенеративно-дистрофические процессы в костной ткани, лежащие в основе болезни Келера, послужили основанием для ее причисления к группе остеохондропатий, куда также входят болезнь Кальве, болезнь Тиманна и болезнь Шляттера. Болезнь Келера встречается в основном в детском возрасте. Наиболее часто болезнь Келера I наблюдается у мальчиков от 3 до 7 лет, а болезнь Келера II — у девочек 10-15 лет.
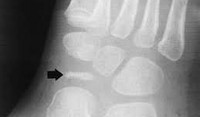
Болезнь Келера
Симптомы
Симптомы болезни Келера I.
Болезнь Келера I характеризуется появлением на тыльной стороне стопы ближе к ее внутреннему краю припухлости, обусловленной отечностью тканей этой области. Отсутствие покраснения кожи и местного повышения температуры в области отека свидетельствует в пользу невоспалительного характера происходящих изменений. Отмечается болезненность пораженной области при прощупывании и при нагрузке на стопу, утомляемость ребенка при ходьбе. Чтобы избежать боли при ходьбе, дети, имеющие болезнь Келера, ставят ногу с упором на наружный край стопы. Может наблюдаться хромота. Со временем боль усиливается и приобретает постоянный характер, не исчезая даже при полном покое. Болезнь Келера I длится в среднем около года и может привести к стойкой деформации ладьевидной кости.
Симптомы болезни Келера II.
Болезнь Келера II проявляется припухлостью и болезненностью в области пораженной плюсневой кости. Чаще всего встречается поражение II и III плюсневых костей. Возможен двусторонний характер патологических изменений. При этом симптомы воспаления не наблюдаются. Болезнь Келера II начинается с появления неинтенсивного болевого синдрома, поначалу проявляющегося лишь при нагрузке на передние отделы стопы. Характерно усиление боли при прощупывании пораженной области и во время ходьбы, особенно по неровному грунту или в обуви со слишком тонкой и мягкой подошвой. Со временем пациенты жалуются на то, что боль в стопе становиться постоянной, более интенсивной и сохраняется даже в покое. Отмечается укорочение пальца, который примыкает к головке подвергшейся некрозу плюсневой кости. Объем движений в суставе, сформированном пораженной плюсневой костью, ограничивается. Болезнь Келера II протекает в среднем в течение 2-3 лет.
Боль в голеностопе.
Диагностика
Диагностика болезни Келера I.
Болезнь Келера I диагностируется рентгенологически. На рентгенограммах стопы в начале заболевания отмечается остеопороз ладьевидной кости, вызванный асептическим разрушением ее губчатого вещества. Затем выявляется уплотнение точек окостенения, сплющивание и уплотнение ладьевидной кости. Еще позже наблюдается дефрагментация ладьевидной кости, т. Е. Ее распад на отдельные костные фрагменты в результате прогрессирования некротического процесса.
Диагностика болезни Келера II.
Диагностика заболевания основана на рентгенологическом исследовании стопы, в ходе которого выявляются патологические изменения в головке пораженной плюсневой кости. В зависимости от срока заболевания может наблюдаться остеопороз, уплотнение и деформация головки плюсневой кости, ее патологический перелом и дефрагментация.
Причины
Как и этиология других остеохондропатий факторы, вызывающие болезнь Келера, пока окончательно не изучены. Большинство исследователей склонны считать основной причиной некротических изменений костной ткани нарушение ее питания за счет расстройства местного кровоснабжения. Нарушения васкуляризации кости в свою очередь могут быть обусловлены врожденными особенностями кровообращения этой области, наличием поперечного или продольного плоскостопия, ношением неудобной или слишком тесной обуви, повторными травмами: ушибами, подвывихами или вывихами стопы, переломами костей стопы Роль благоприятствующих факторов в развитии болезни Келера могут играть различные обменные нарушения и эндокринопатии (гипотиреоз, сахарный диабет, ожирение).
Профилактика
Предупредить болезнь Келера у ребенка поможет правильный подбор обуви, которая должна соответствовать размеру ноги, быть удобной и не слишком жесткой. Следует избегать травм стопы, а при их получении незамедлительно обращаться к травматологу и следовать всем его рекомендациям. Поскольку болезнь Келера связана с плоскостопием, то его своевременное лечение также имеет профилактическое значение.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
 Лиссэнцефалией (буквально «гладкий мозг») называется весьма большая по объему группа нарушений в развитии головного мозга, которая характеризуются частичным отсутствием или же плохим развитием извилин, расположенных в полушариях головного мозга.
Лиссэнцефалией (буквально «гладкий мозг») называется весьма большая по объему группа нарушений в развитии головного мозга, которая характеризуются частичным отсутствием или же плохим развитием извилин, расположенных в полушариях головного мозга.
В итоге поверхность мозга становится удивительно гладкой. Этот дефект начинает проявляться еще с 12 недели развития плода, и заканчивает формироваться спустя 2 недели, и вызван он нарушенным процессом передвижения нейронов в этот период.
Головной мозг, в нормальном состоянии у каждого здорового человека, имеет множество складок и борозд. У плода с этим отклонением, структура развивается не своеобразным способом, у таких пациентов складки и борозды либо частично отсутствуют, либо их вовсе нет, и произошедшие изменения уже не обратимы.
Точно так же, как и многие другие дефекты в развитии головного мозга, лиссэнцефалия берет под контроль большой спектр фенотипов, они могут колебаться в соответствии с тяжестью от переменной агирии, либо до полной.
Сопутствующие заболевания и нарушения
Сопутствовать данному пороку развития может — синдром Миллера-Дикера, либо Уокера-Варбурга.
Синдром Миллера-Дикера характеризуется классической лиссэнцефалией, при таком синдроме под воздействие аномалии попадают лицевые

Синдром Уокера-Варбурга
мышцы и проявляются другие возможные дефекты, которые можно встретить у людей только с таким синдромом.
Особенностью синдрома является потеря в хромосоме 17 нескольких генов. За лиссэнцефалию отвечает потеря гена PAFAH1B1, это доказанный учеными факт.
А вот при утере гена YWHAE, лиссэнцефалия может усложниться. Последствия после потери других генов при данном синдроме еще не известны.
Синдром Уокера-Варбурга – очень редкая врожденная дистрофия мышц, связанная с нарушением работы головного мозга и лицевых мышц.
Классификация заболевания
На данный момент существует большое количество систем для классификации патологии. Рассмотрим наиболее популярную в нынешних реалиях, в которой имеется, — тип первый – классический, и тип второй – «булыжник».
Классический тип мутации
Развитие патологии происходит из-за мутировавшего гена PAFAH1B1. Такой дефект можно разделить на изолированную лиссэнцефалию и синдром Миллера-Дикера.
Изолированная патология проходит без развития других дефектов в геноме, во втором случае наблюдается развитие сопутствующих аномалий из-за мутации в DCX гене.
Тип «булыжник»
Второй тип «булыжник» включает следующие типа аномалии:
- заболевание – мышцы-глаз-мозг;
- синдромы Уокера-Варбурга и Фукуямы.
Кроме этого, существуют также другие подтипы:
- микролиссэнцефалия;
- синдром Нормара-Робертса;
- патология вызванная мутировавшим ARX геномом.

Типы развития
Рассмотрим патологию, базируясь на 4 типах развития.
Тип первый
Мозг ребенка можно сравнить с мозгом плода на 23 неделе развития. Кора состоит из четырех слоев нейронов, развивается пахигирия, агирия.
Особенности:
- гладкая наружная часть коры мозга;
- сильно уменьшен объем белого вещества в мозгу;
- ленточная гетеротопия, которая отделена от коры белой полосой;
- гипоплазия ствола.
Тип второй
Кора мозга значительно толще, чем должна быть, без нормального слоевого строения, имеются нарушения в сосудистом древе; присутствует  гидроцефалия.
гидроцефалия.
Особенности:
- гладкая поверхность коры;
- гипоплазия;
- размыта граница между белым и серым веществом;
- утолщение коры;
- агенезия мозолистого тела;
- у некоторых больных в затылочной области – энцефалоцеле.
Тип третий
Особенности:
- белое вещество в меньших количествах, нежели нужно;
- гипоплазия;
- начинают выделяться извилины, но это малозаметно.
Тип четвертый
Особенности:
- остановка миелинизации;
- головной мозг очень мал в размере;
- количество нейронов составляет 35% нормы;
- на коре виднеются некоторые извилины и борозды с нормальной толщиной.
В некоторых случаях используют еще одну классификацию лиссэнцефалии, ее основывают в зависимости от тяжести болезни:
- самый тяжелый класс, тотальная агирия, нет никаких извилин;
- имеется небольшое количество складок на затылочных и лобных полосках, диффузная агирия;
- сочетание пахигирии и агирии;
- диффузная пахигирия;
- пахигирия спереди и гетеротопия;
- гетеротопия под корой.
Первая и четвертая степень очень редка, вторая имеется у детей с синдромом Миллера-Дикера. Самое частое — сочетание в третьей степени, обычно такая патология состоит из задней агирии и лобной пахигирии.
Тип первый, то есть классический, встречается у 12 человек на миллион рожденных.
Видео по теме:
Причины и патогенез аномального развития
 Определить из-за чего начал формироваться дефект — задача не из легких, так как их причин быть целое множество. Примерно у 60% людей с лиссэнцефалией замечены мутация в гене LIS1.
Определить из-за чего начал формироваться дефект — задача не из легких, так как их причин быть целое множество. Примерно у 60% людей с лиссэнцефалией замечены мутация в гене LIS1.
Еще у примерно 3% людей, конкретно с классическим типом патологии, можно отметить мутацию в гене TUBA1A. Примерно у 30% пациентов с гипоплазией наблюдаются изменения в TUBA1A гене.
Иные причины возникновения могут быть самыми разными, например, при развитии инфекционных заболеваний в матке в первом триместре беременности, или же на ранних сроках у плода было плохое кровоснабжение в области мозга, в результате чего начала развиваться лиссэнцефалия.
Процесс развития коры головного мозга:
- нервные клетки начинают делиться;
- кора дифференцируется;
- нейроны движутся из матрикса.
Хронология развития плода в норме должна происходить таким образом:
- на 11-12 неделях начинают формироваться оба полушария;
- на 16 – начинают формироваться борозды и извилины;
- на 20 — уплотняется кортикальное плато, начинают формироваться первичные и центральные борозды;
- на 23-26 — сформировываются ольфакторные борозды;
- на 28 неделе развития внутри утробы матери, начинают формирование борозды в височной области.
Кора головного мозга формируется за счет миграции мозговых клеток – нейронов, которые находятся в разных слоях мозга. В процессе их передвижения, клетки, которые сформировались на более позднем этапе, передвигаются дальше, чем предыдущие, в результате происходит формирование нового слоя. Этот процессе регулируется специальными белками N-кадгерином и рилоном.
В норме, все основные борозды и извилины начинают свое формирование после окончания передвижения нейронов, именно нарушение данного процесса влияет на неправильное создание извилин и борозд.
Постановка диагноза и возможности медицины
Обычно в таком случае диагноз ставится при рождении или немного позже, после проведения УЗИ и исследования результатов КТ или МРТ.
Но врачи могут заподозрить наличие патологии еще во время беременности, ведь они регулярно проводят УЗИ плода, и постоянно изучают его состояние, в частности и развитие головного мозга.
Если врач заподозрит лиссэнцефалию, кроме УЗИ нужно будет проводить еще несколько методов диагностики, например, анализ на явность мутирующих генов и ЯРМ-томографию.
Обнаружить данную патологию можно не раньше, чем на 20 неделе беременности, поскольку поверхность мозга до этого времени, и без каких-либо нарушений, является гладкой. Именно после данного периода, можно заметить отклонения в развитии плода.
Вылечить данный порок невозможно, есть возможность симптоматического лечения, и оно зависит от стадии развития и места расположения дефектов. Необходимо поддерживать постоянный уход.
Для людей с гидроцефалией обычно делают шунтирование, а для контроля судорог принимаются специальные лекарственные препараты.
Прогноз продолжительности жизни
Главным фактором, который определяет прогноз продолжительности жизни ребенка с диагнозом лиссэнцефалия, является степень тяжести заболевания. Так некоторые из пациентов имеют более-менее нормальное интеллектуальное развитие.
В основном дети, с более тяжелой формой патологии, не способны прожить дольше 10 лет, но даже дожив до этого возраста, их развитие все равно остается на уровне 4-6 месячного ребенка.
Главной причиной смерти больных, в основном, является связь с аспирацией питательных продуктов или жидкости, с очень тяжелыми приступами, или с болезнями дыхательных путей.
Источник