Синдром марфана только по наследству

Классический синдром Марфана (болезнь MFS) это наследственное заболевание соединительной ткани, передающееся аутосомно-доминантным путем.
Откуда стало известно о синдроме Марфана?
Вот любопытное сопоставление двух классических факторов. Первый фактор: классическая эпидемиология сосудистых заболеваний. Второй: классический тип аневризмы аорты (как мы уже знаем, это аневризм ее грудного участка). Итак, классическая эпидемиология распределяет причины аневризмы грудного участка аорты следующим образом:
- в 80 процентах случаев – атеросклероз и повышенное артериальное давление;
- в 20 процентах случаев – два врожденных заболевания: либо синдром Марфана, либо порок аортального клапана, затрудняющий нагнетание крови из левого сердечного желудочка в аорту.
В последние годы в реестр классической эпидемиологии внесены значительные коррективы. Так, наиболее распространена сейчас аневризма не грудного, а брюшного участка аорты. Детализированы и причины данного нарушения в строении главной артерии. Но вот что интересно: атеросклероз и гипертония по-прежнему обусловливают 80 процентов всех случаев аневризмы. А оставшиеся 20 процентов поделены между синдромом Марфана, пороком аортального клапана и еще целым рядом генетических заболеваний, о которых в недавнем прошлом медицина практически не имела представления.
Опасным возрастом признана середина четвертого десятка. Если у человека к 35 годам проявляются признаки аневризмы аорты, то это, скорей всего, результат генетического заболевания, о котором он, наверное, и не ведал.
Точно знать причину сосудистого заболевания – всегда важно. В случае же с торакальной аневризмой аорты (то есть с поражением ее грудного участка) выявление генетической причины приобретает особую важность, поскольку от этого зависит прогноз дальнейшей прогрессии аневризмы, риска разрыва или расслоения аорты. Зависит, естественно, и выбор лечебных средств: радикальных (хирургических) или паллиативных.
Каковы же причины синдрома Марфана
Аутосомно-доминантный путь наследования исходит от одного из родителей (а не от обоих), наследуемый признак присутствует не у всех его детей, а у половины потомства.
Разница доминантных и рецессивных путей
Если речь идет о наследственном заболевании, то его причина коренится в передаче патологической мутации генов. Родитель, который аутосомно-доминантным путем передает потомству мутированный ген, является не только его носителем – он к тому же и болен этой болезнью! И та половина потомства, которой передалась мутация, тоже больны.
Для сравнения: при аутосомно-рецессивной передаче наследственной болезни носителями одной и той же генетической мутации являются оба родителя – при том, что болезни как таковой у них нет! Однако четверти их потомства передается именно болезнь – хотя больной не передает эту мутацию своему потомству. А половине потомства передается, наоборот, способность передавать собственному потомству мутированный ген, хотя сами они болезнью, происходящей от этой мутации, не болеют. Вспомним пример британской королевы Виктории, которая передала гемофилию мужскому потомству почти всех монархических домов Европы (в том числе российскому цесаревичу Алексею). Но сама гемофилией не болела.
Марфан классический и неклассический
Вернемся, однако, к синдрому Марфана. При классической его форме всегда необходимо считаться с тем, что кто-то из родителей больного тоже имеет (или имел) это заболевание.
Известны и неклассические формы, когда болезнь развивается из-за мутировавшего гена без признаков наследования. Иными словами, мутация не перешла от отца или матери, она произошла в генах самого пациента.
«Самомутировавший» ген является причиной 25-40 процентов всех заболеваний с признаками синдрома Марфана. В остальных случаях сказывается наследственное происхождение. В обоих формах болезнь встречается в среднем у одного человека на десять тысяч населения.
Источник
Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
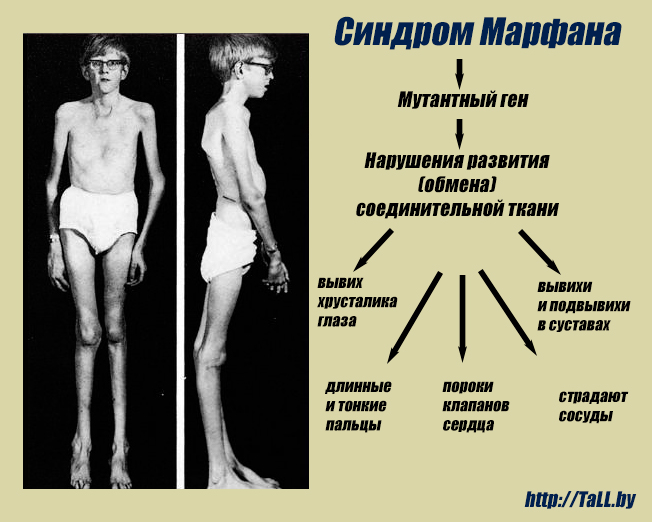
Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.

Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.

Симптомы синдрома Марфана
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз, сколиоз;
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— плоскостопие;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— высокое нёбо;
— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.

Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:

Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн. Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Источники и ссылки:
1. Причины высокого роста — статья, в которой рассмотрены основные причины высокого роста человека.
2. Синдром Марфана в википедии — о синдроме Марфана в свободной энциклопедии.
3. Русскоговорящее сообщество людей с синдромом Марфана — форум о синдроме Марфана, общение, обсуждение болезни.
4. Международный фонд синдрома Марфана — фонд помощи больным с синдромом Марфана.
5. Группа вконтакте для больных синдромом Марфана.
Источник
Синдром Элерса-Данло — это группа наследственных заболеваний, известных также как «гиперэластичность кожи». Болезнь возникает из-за генетических мутаций, приводящих к дефициту выработки коллагена. Вследствие такого недостатка человек, страдающий синдромом, обладает гиперподвижностью суставов. То есть пальцы, руки и ноги в буквальном смысле могут гнуться в обратную строну. И самое странное, что этот синдром, по-видимому, как-то связан с генетическим кодом, присущим именно русскому человеку.
Гнется, как на шарнирах
Болезнь была впервые выявлена около столетия назад двумя учеными-медиками, в честь которых синдром и получил название. Как оказалось, заболевание весьма разнообразно и имеет несколько типов — от относительно распространенных (1 случай на 10—15 тыс. человек) до крайне редких (несколько десятков случае во всем мире). Об одном из последних и пойдет речь.
Так называемый синдром Элерса-Данло с кифосколиозом — одна из разновидностей заболевания, выявленная относительно недавно. Предположение о существовании такой формы синдрома было высказано еще в 2012 году. Одними из первых это сделали медики из США П. Бейтон, Р. Грэйхем и Г. Берд в своей работе «Гипермобильность суставов». Но лишь спустя 6 лет удалось обработать все данные по анализам ДНК и выделить болезнь из общей группы.
Синдром Элерса-Данло с кифосколиозом возникает из-за нарушений выработки коллагена. Этот белок составляет основу соединительной ткани человека, из которой состоят хрящи, суставы, нижний слой кожи. В результате заболевания суставы человека приобретают способность гнуться под самыми невообразимыми углами, что часто приводит к различным травмам, а кожа у людей с синдромом может очень растягиваться.
В настоящее время известно около 60 случаев болезни во всем мире. Установлено, что она вызвана мутацией гена FKBP14, и более половины всех случаев проявления такой формы синдрома зарегистрировано в России. Ученые предполагают, что мутация гена FKBP14 каким-то образом связана с генотипом именно русских людей (и более крупной группы восточных славян), но пока точная взаимосвязь не установлена. Например, сотрудница Института общей генетики РАМН им. Н. И. Вавилова, кандидат биологических наук Светлана Боринская, в своей работе «Зависят ли наши болезни от национальности?» высказывает предположение о взаимосвязи мутации с исторически сложившимся пищевым рационом русского народа.
Синдром Марфана и талант
Следует отметить, что мутации гена FKBP14 являются причиной и другого заболевания (по-видимому, связанного с синдромом Элерса-Данло) — синдрома Марфана. Эта болезнь также характеризуется нарушениями структуры и регенерации соединительной ткани, из-за чего скелет больного становится хрупким, а суставы приобретают гиперподвижность. Носители синдрома Марфана отличаются от здоровых людей внешне: они высокие и обычно довольно худые, обладают непропорционально длинными руками и ногами, крупными кистями и большим носом.
Страдающие синдромом Марфана обычно имеют патологии зрения и внутренних органов, но самое интересное, что среди них чаще, чем в среднем, встречаются талантливые люди. Так, носителями синдрома были Корней Чуковский, Ханс Кристиан Андерсен, Никола Паганини, Авраам Линкольн.
Зависят ли умственные способности от гена FKBP14, могут ли среди больных синдромом Элерса-Данло с кифосколиозом или их детей быть гении и как этот ген связан с национальностью? Пока на эти вопросы нет однозначных ответов.
Видео дня. Мадонна пришла на митинг в защиту чернокожих на костылях
Источник
Синдром Элерса-Данло — это группа наследственных заболеваний, известных также как «гиперэластичность кожи». Болезнь возникает из-за генетических мутаций, приводящих к дефициту выработки коллагена. Вследствие такого недостатка человек, страдающий синдромом, обладает гиперподвижностью суставов. То есть пальцы, руки и ноги в буквальном смысле могут гнуться в обратную строну. И самое странное, что этот синдром, по-видимому, как-то связан с генетическим кодом, присущим именно русскому человеку.
Гнется, как на шарнирах
Болезнь была впервые выявлена около столетия назад двумя учеными-медиками, в честь которых синдром и получил название. Как оказалось, заболевание весьма разнообразно и имеет несколько типов — от относительно распространенных (1 случай на 10—15 тыс. человек) до крайне редких (несколько десятков случае во всем мире). Об одном из последних и пойдет речь.
Так называемый синдром Элерса-Данло с кифосколиозом — одна из разновидностей заболевания, выявленная относительно недавно. Предположение о существовании такой формы синдрома было высказано еще в 2012 году. Одними из первых это сделали медики из США П. Бейтон, Р. Грэйхем и Г. Берд в своей работе «Гипермобильность суставов». Но лишь спустя 6 лет удалось обработать все данные по анализам ДНК и выделить болезнь из общей группы.
Синдром Элерса-Данло с кифосколиозом возникает из-за нарушений выработки коллагена. Этот белок составляет основу соединительной ткани человека, из которой состоят хрящи, суставы, нижний слой кожи. В результате заболевания суставы человека приобретают способность гнуться под самыми невообразимыми углами, что часто приводит к различным травмам, а кожа у людей с синдромом может очень растягиваться.
В настоящее время известно около 60 случаев болезни во всем мире. Установлено, что она вызвана мутацией гена FKBP14, и более половины всех случаев проявления такой формы синдрома зарегистрировано в России. Ученые предполагают, что мутация гена FKBP14 каким-то образом связана с генотипом именно русских людей (и более крупной группы восточных славян), но пока точная взаимосвязь не установлена. Например, сотрудница Института общей генетики РАМН им. Н. И. Вавилова, кандидат биологических наук Светлана Боринская, в своей работе «Зависят ли наши болезни от национальности?» высказывает предположение о взаимосвязи мутации с исторически сложившимся пищевым рационом русского народа.
Синдром Марфана и талант
Следует отметить, что мутации гена FKBP14 являются причиной и другого заболевания (по-видимому, связанного с синдромом Элерса-Данло) — синдрома Марфана. Эта болезнь также характеризуется нарушениями структуры и регенерации соединительной ткани, из-за чего скелет больного становится хрупким, а суставы приобретают гиперподвижность. Носители синдрома Марфана отличаются от здоровых людей внешне: они высокие и обычно довольно худые, обладают непропорционально длинными руками и ногами, крупными кистями и большим носом.
Страдающие синдромом Марфана обычно имеют патологии зрения и внутренних органов, но самое интересное, что среди них чаще, чем в среднем, встречаются талантливые люди. Так, носителями синдрома были Корней Чуковский, Ханс Кристиан Андерсен, Никола Паганини, Авраам Линкольн.
Зависят ли умственные способности от гена FKBP14, могут ли среди больных синдромом Элерса-Данло с кифосколиозом или их детей быть гении и как этот ген связан с национальностью? Пока на эти вопросы нет однозначных ответов.
Видео дня. Как быстро избавиться от пяточной шпоры
Источник