Синдром луи бар атаксия телеангиэктазия


Синдром Луи-Бар (атаксия-телеангиэктазия) — наследственное заболевание, проявляющееся мозжечковой атаксией, телеангиэктазиями кожи и конъюнктивы глаз, недостаточностью Т-клеточного звена иммунитета. Последнее приводит к тому, что синдром Луи-Бар сопровождается частыми респираторными инфекциями и склонностью к возникновению злокачественных опухолей. Диагностируется синдром Луи-Бар на основании анамнеза и клинической картины заболевания, данных иммунограммы, результатов офтальмологического и отоларингологического обследования, МРТ головного мозга и рентгенографии легких. В настоящее время синдром Луи-Бар не имеет специфического и эффективного лечения.
Общие сведения
Синдром Луи-Бар впервые был описан в 1941 году во Франции. Нет точных данных о том, с какой частотой синдром Луи-Бар встречается среди современного населения. По некоторым сведениям эта цифра составляет 1 случай на 40 тысяч новорожденных. Однако, необходимо учитывать, что при смерти в раннем детском возрасте синдром Луи-Бар обычно остается не диагностированным. Известно, что заболевание одинаково часто поражает мальчиков и девочек. В неврологии синдром Луи-Бар относится к так называемым факомотозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа—Вебера, туберозный склероз и др.
Синдром Луи-Бар
Причины и патогенез синдрома Луи-Бар
В основе патологических изменений, сопровождающих синдром Луи-Бар, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т. е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей.
Морфологически атаксия-телеангиэктазия характеризуется дегенеративными изменениями тканей мозжечка, в частности потерей зернистых клеток и клеток Пуркинье. Дегенеративные изменения могут затрагивать зубчатое ядро мозжечка (nucleus dentatus), черную субстанцию (substantia nigra) и некоторые отделы коры головного мозга, иногда поражаются спиномозжечковые пути и задние столбы спинного мозга.
Синдром Луи-Бар сочетается с гипоплазией или аплазией тимуса, а также с врожденным дефицитом IgA и IgE. Эти нарушения в системе иммунитета приводят к появлению у пациентов частых инфекционных заболеваний, склонных к длительному и осложненному течению. Кроме того, иммунные нарушения могут потенцировать развитие злокачественных новообразований, зачастую берущих свое начало в структурах лимфоретикулярной системы.
Клинические проявления синдрома Луи-Бар
Атаксия. Наиболее часто синдром Луи-Бар начинает проявляться клинически в возрасте от 5 месяцев до 3 лет. Во всех случаях заболевания синдром Луи-Бар манифестирует с появления мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание во время двигательного акта (интенционный тремор), качание туловища и головы. Зачастую атаксия настолько выражена, что имеющий синдром Луи-Бар больной не может ходить. Мозжечковая атаксия сочетается с мозжечковой дизартрией, характеризующейся невнятной скандированной речью. Отмечается мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие.
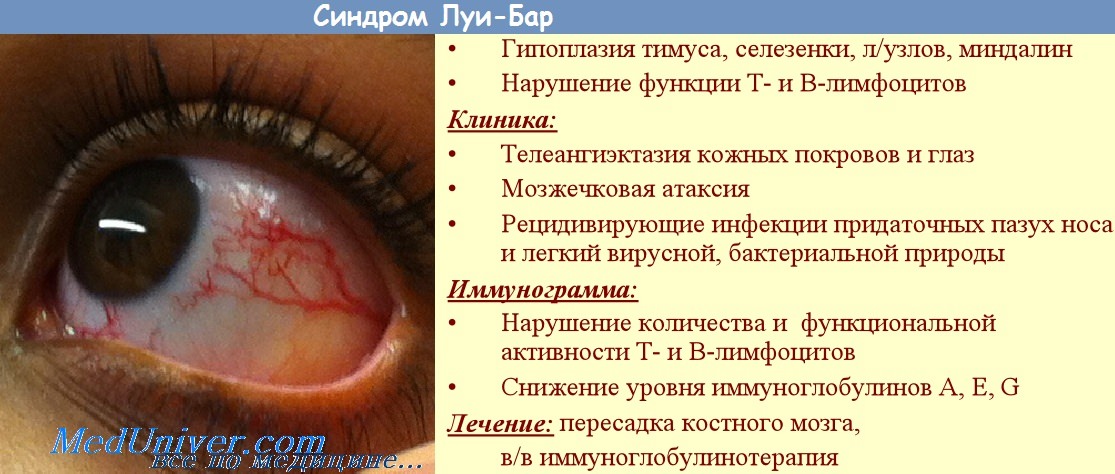
Телеангиэктазии. В большинстве случаев появление сопровождающих синдром Луи-Бар телеангиэктазий происходит в возрасте от 3 до 6 лет. В некоторых случаях их возникновение отмечается в более поздний период и очень редко в течение первого месяца жизни. Телеангиэктазии (сосудистые звездочки) представляют собой имеющие различную форму красноватые или розовые пятнышки или разветвления. Они обусловлены расширением мелких сосудов кожи. Следует отметить, что телеангиэктазии могут быть проявлением многих других заболеваний (например, розацеа, СКВ, дерматомиозита, пигментной ксеродермы, хронического лучевого дерматита, мастоцитоза и пр.). Однако в сочетании с атаксией они дают специфическую для синдрома Луи-Бар клиническую картину.
Синдром Луи-Бар характеризуется изначальным возникновением телеангиэктазий на конъюнктиве глазного яблока, где они имеют вид «паучков». Затем сосудистые звездочки появляются на коже век, носа, лица и шеи, локтевых и коленных сгибов, предплечий, тыльной поверхности стоп и кистей. Телеангиэктазии могут также наблюдаться на слизистой оболочке мягкого и твердого неба. Наиболее выражены сосудистые звездочки в тех местах кожного покрова, где он подвергается воздействию солнечных лучей. В первую очередь это лицо, где телеангиэктазии образуют целые «пучки». При этом кожа теряет свою эластичность и становится плотной, что напоминает изменения, типичные для склеродермии.
Кожные проявления атаксии-телеангиэктазии могут включать появление веснушек и пятен цвета кофе с молоком, участков обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Может наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне или проявления псориаза.
Инфекции дыхательных путей. Характеризующее синдром Луи-Бар поражение иммунной системы приводит к возникновению частых рецидивирующих инфекций дыхательных путей и уха: хронических ринитов, фарингитов, бронхитов, пневмоний, отитов, синуситов. Их особенностями являются: стертость границ между периодом обострения и ремиссии, скудность физикальных данных, плохая чувствительность к антибактериальной терапии и длительное течение. Каждая подобная инфекция может стать для больного атаксией-телеангиэктазией смертельно опасной. Частые заболевания легких приводят к развитию бронхоэктазов и пневмосклероза.
Злокачественные новообразования. Среди пациентов, имеющих синдром Луи-Бар, злокачественные опухолевые процессы отмечаются в 1000 раз чаще, чем в среднем у населения. Наиболее распространенными среди них являются лейкемия и лимфома. Особенностью онкопатологии в случае синдрома Луи-Бар является повышенная чувствительность пациентов к воздействию ионизирующего излучения, что полностью исключает применение лучевой терапии при их лечении.
Диагностика синдрома Луи-Бар
Постановка диагноза атаксии-телеангиэктазии требует комплексного подхода, учитывающего анамнез заболевания, его клинические проявления, данные иммунологических и инструментальных исследований, а также результаты ДНК-диагностики. Пациент с подозрением на синдром Луи-Бар должен пройти обследование не только у невролога, но и у дерматолога, отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога.
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у 1/3 пациентов наблюдается снижение количества лимфоцитов. Обязательно проводится исследование уровня иммуноглобулинов крови, которое выявляет значительное снижение IgA и IgE, в 10-12% случаев IgG. Примерно у 40% пациентов синдром Луи-Бар сопровождается аутоиммунными реакциями, о которых свидетельствует наличие аутоантител к митохондриям, тиреоглобулину, иммуноглобулинам.
Из инструментальных способов диагностики синдрома Луи-Бар могут применяться: УЗИ тимуса, МРТ головного мозга, фарингоскопия, риноскопия, рентгенография легких. При помощи УЗИ диагностируется аплазия или гипоплазия тимуса. МРТ головного мозга выявляет атрофию мозжечка, расширение IV желудочка. Рентгенография легких необходима для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений.
Синдром Луи-Бар следует дифференцировать с атаксией Фридрейха, болезнью Рандю-Ослера, атаксией Пьера-Мари, болезнью Гиппеля-Линдау и др.
Лечение и прогноз синдрома Луи-Бар
К сожалению, эффективные методы лечения синдрома Луи-Бар до настоящего времени остаются предметом поиска. В современной медицине возможно применение лишь паллиативного симптоматического лечения соматических и иммунологических нарушений. Продлению жизни пациентов, имеющих синдром Луи-Бар, способствует иммунокоррегирующая терапия препаратами тимуса и гамма-глобулином, витаминотерапия в высоких дозировках и интенсивная терапия любого инфекционного процесса. По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
В связи с отсутствием эффективных способов лечения синдром Луи-Бар имеет неблагоприятный прогноз как для выздоровления, так и для жизни. Больные этим заболеванием редко доживают до 20 лет. В большинстве случаев они погибают от инфекционных осложнений и онкологических заболеваний.
Источник
Предрасположенность организма к частым инфекционным заболеваниям, злокачественным или доброкачественным новообразованиям называется синдром Луи Бара. Довольно редкое, но вместе с тем очень опасное заболевание, передается по наследству и встречается один раз на 40 тысяч человек. Однако, это цифра довольно условная, так как заболевание не всегда удается диагностировать. Так, в раннем младенческом возрасте малыш может погибнуть от данного недуга, но причина так и останется не выясненной.
Общие сведения
Данное заболевание впервые было диагностировано в 1941 году французским врачом Луи Бар. Болезнь является аутосомно-рецессивным заболеванием.
Аутосомно-рецессивное – значит проявляющееся в случае наличия болезни у обоих родителей.
Синдром Луи Барра заключается в поражении Т-звена иммунной системы, что в итоге приводит к неправильному ее формированию. Результатом является частые возникновения инфекционных заболеваний у ребенка, причем с каждой новой болезнью ее тяжесть увеличивается, что сказывается на последствия и общее состояние малыша. В дальнейшем (иногда и параллельно с инфекциями) у младенца могут расти новообразования (чаще злокачественные).
Как правило, больного ребенка видно, так как в ходе заболевания у пациента образуются кожные нарушения, появляется неровность походки (в результате поражения мозжечка), отставание в развитии.
Причины развития недуга
Как было сказано ранее, синдром Бара является наследственным заболеванием и передается только по наследству. Если из родителей только один имеет хромосомные нарушения, ребенок приобретет данный недуг с 50% вероятностью, ну а если оба родителя – то вероятность болезни малыша 100%.
В настоящее время уровень диагностики довольно велик и позволяет выявить возможные проблемы еще на этапе формирования зародыша, однако, данный синдром коварен и часто доктор лишь делает предположение о том, что ребенок может приобрести и дает примерный процент, что обнадеживает будущую маму.
Глазные проявленияДля того, чтобы не мучить себя подобными переживаниями достаточно знать, какие фактор оказывают негативное воздействие при развитии синдрома, в том числе:
- вредные привычки во время беременности (курение, злоупотребление алкоголем);
- частые стрессы будущей мамы;
- внешнее воздействие (ядовитые вещества, радиоактивное излучение).
Симптомы недуга
Как и любая другая болезнь, синдром Луи Бара имеет свои отличительные особенности, так у больных могут проявляться следующие симптомы:
- мозжечковая атаксия;
- телеангиэктазия;
- инфекционная предрасположенность;
- новообразования.
Мозжечковая атаксия
Данный симптом проявляется практически с первых месяцев жизни, но невооруженным взглядом становиться заметным в тот период, когда малыш начинает учиться ходить. В ходе поражения мозжечка у ребенка развивается нетвердость походки. В более тяжелых формах малыш не может самостоятельно передвигаться или даже стоять.
Проявления на лицеКроме того, у больного могут развиться косоглазие, глазодвигательные проблемы, нистагм, у больного могут пропасть или снизиться сухожильные рефлексы. Кроме того, в результате болезни может развиться мозжечковая дизартрия, которая проявляется в виде невнятной речи.
Дизартрия – ограничение подвижности органов речи (нёбо, язык, губы).
Телеангиэктазия
Данный симптом является менее опасным, чем предыдущий, но может причинять малышу некоторые неудобства. Телеангиэктазия – означает наличие на коже расширенных капилляров, которые выглядят в виде розовых либо красных звездочек или паучков. Как правило, звездочки из кровеносных капилляров начинают образовываться к 3-6 годам жизни малыша.
Наиболее распространенные места формирования:
- глазное яблоко;
- конъюнктивы глаз (слизистая глаза за нижним веком);
- уши;
- нос;
- тыльная поверхность стоп;
- места сгибов (локтевые впадины, коленные впадины, подмышечные впадины).
В самом начале телеангиэктазия проявляется на конъюнктивах глаз, после чего страдает кожа лица и постепенно опускается ниже по телу. Имели место случаю формирования подобных «звездочек» на мягком нёбе.
Помимо прочего, к кожным высыпаниям при синдроме Луи Бара относят веснушки, сухость кожи, ранняя седина волос (в случае с маленькими детьми это особенно заметно).
Инфекционная предрасположенность
Любой ребенок болеет, но, что касается синдрома Луи Бар это происходит аномально часто и с каждым разом тяжесть этих заболеваний усиливается, но любая инфекция может вызвать смерть больного.
Кожные проявленияКак правило, недуг вызывает возникновение только дыхательных и ушных инфекций (ринит, фарингит, бронхит, отит, синусит).
Стоит отметить, что лечению такие инфекции поддаются хуже, чем обычные заболевания, что и обуславливает довольно долгий процесс излечения.
Новообразования
Как правило, при наличии синдрома Бара у больного в 1000 раз больше шансов развития онкологических новообразований злокачественного типа. Наиболее часто встречающиеся из них – лейкемия и лимфома.
Главная трудность связанная с лечением таких больных заключается в невозможности применять лучевую терапию, в связи с гиперчувствительностью пациентов к ионизирующему облучению.
Диагностика
Для постановки диагноза не достаточно клинических проявлений, так как многие симптомы данной болезни характерны и для других
недугов. Как правило, требуется консилиум врачей, в который входят:
- дерматолог;
- отоларинголог;
- офтальмолог;
- иммунолог;
- пульмонолог;
- онколог;
- невролог.
Помимо прочего больному назначают следующие анализы:
- клинический анализ крови;
исследование иммуноглобулина. Что может быть в ушной раковине при данном недуге
Инструментальная диагностика включает:
- ультразвуковое (УЗИ) тимуса;
Тимус – или вилочковая железа, орган в котором зреют иммунные Т-клетки организма
- магнитно-резонансная томография (МРТ);
- фарингоскопия;
- риноскопия;
- рентгенография легких.
При расшифровке анализов крови возможно низкое количество лимфоцитов. При исследовании иммуноглобулина обычно отмечается снижение IgA и IgE.
IgA и IgE – титры антител уровня А отвечают за местный иммунитет, а Е за аллергические реакции.
Кроме того, возможно обнаружение в крови аутоантител к митохондриям, тиреоглобулину и иммуноглобулину.
Аутоантитела – агрессивные антитела, атакующие собственные антитела
Митохондрии – участвуют в процессе формирования энергии
Тиреоглобулин – белок, предшественник гормона щитовидной железы, выявляется в крови большинства здоровых людей
Лечение
Лечение синдрома Луи Бара в настоящее время является открытым вопросом и эффективного способа устранения данного недуга еще не существует. Основу терапии составляет устранение возникающих симптомов и продление жизни больным.
Так, при лечении применяют:
- Противовирусные препараты.
- Широкие антибиотики.
- Противогрибковые средства.
- Глюкокортикостероиды.
Так как инфекционные заболевания плохо поддаются лечению, больному показано применение комплекса витаминов в большой дозировке, для стимуляции собственных иммунных резервов.
Прогноз
В связи с отсутствием эффективного лечения максимальный срок жизни пациентов, с диагнозом синдром Луи Бара не превышает 20 лет. Однако, даже до такого возраста доживают лишь единицы. Злокачественные новообразования, серьезные инфекционные заболевания убивают больных намного раньше.
Итак, пока врачи не научились лечить такие редкие и опасные болезни риск заболеть существует у каждого. Ну а молодые мамы в ответе за своих еще не родившихся детей, и ведение не здорового образа жизни во время беременности – преступление. Берегите себя и своих малышей.
Источник: https://nervivporyadke.ru
Источник
Иммунодефицитный синдром с атаксией-телеангиэктазией. Синдром Луи-Бар
Иммунодефицитный синдром с атаксией-телеангиэктазией [атаксия-телеангиэктазия (AT), синдром Луи-Бар]—наследственный первичный дефицит клеточного и отчасти гуморального иммунитета в сочетании с прогрессирующей мозжечковой атаксией и окулобульбарными телеангиэктазиями.
Этиология синдрома связана с аутосомно-рецессивной наследственностью. R. Peterson и соавт. (1966) высказывали точку зрения, что дефект тимуса, телеангиэктазии и гипоплазия гонад зависят от дефекта мезодермально-мезенхимального зачатка в период эмбриогенеза. Однако этому противоречит вовлечение в процесс ЦНС. Есть точка зрения, что прогрессирующее поражение ЦНС и эндокринных органов обусловлено аутоиммунными процессами, возникающими в результате дефекта тимуса.
W. H. Hitzig (1975) указывает, что у многих больных обнаруживаются в крови аутоантитела. В настоящее время считают, что имеется патогенетическая связь с нарушением функции тимического ретикулоэпителия. Иммунные нарушения зависят от дефекта конечной дифференцировки Т-клеток. Клиника AT характеризуется дефицитом клеточного иммунитета, который, по данным W. H. Hitzig (1975), у 60% больных выражается отсутствием реакции гиперчувствительности замедленного типа и у большинства больных — дефектом лимфоцитов периферической крови, определяемым реакцией с фитогемагглютининами и специфическими антигенами in vitro.
У большинства больных отмечается дефицит IgA, в некоторых случаях описывается, наоборот, увеличение их количества с одновременным уменьшением количества IgG; встречаются также другие варианты дисгаммаглобуминемии. Кроме того, по данным Ю. М. Лопухина, имеет место низкий уровень а-фетопротеина в крови, лимфопения и эозинофилия. Неврологические симптомы болезни вариабельны, у большинства, однако, атаксия развивается с детского возраста в виде нарушений походки, что выявляется обычно до 4 лет жизни. Постепенно атаксия прогрессирует, развивается вторичная мышечная атрофия.
Телеангиэктазии наблюдаются в первый год жизни, иногда позже, на бульбарной конъюнктиве, а затем и в других областях. Больные переживают пубертатный период, но у них не наблюдается развитие вторичных половых признаков. Менструации нерегулярны, у мальчиков прогрессирует атрофия яичек. Отмечается выраженное отставание в физическом развитии. Картину болезни дополняют повторные синопульмональные инфекции. Продолжительность заболевания различна. Иногда больные живут до 39—41 года.
Со стороны лимфоидной ткани обнаруживается значительная гипоплазия преимущественно Т-зависимых зон лимфатических узлов, и селезенки. В солитарных и групповых лимфатических фолликулах желудочно-кишечного тракта отмечается также атрофия с недостаточным формированием фолликулов. В тимусе явления жирового метаморфоза, что не соответствует возрасту.
Дольки представлены очень мелкими островками тимической ткани без телец Гассаля или с единичными образованиями, несколько напоминающими тимические тельца. Кроме того, со стороны эпителия наблюдается появление клеток с гиперхромными ядрами—изменения эти отмечались нами в 2 случаях AT у девочки 10 лет и женщины 24 лет. Со стороны мозжечка — выраженная атрофия с расширением полости IV желудочка.
Микроскопически — тяжелые дистрофические изменения, вплоть до полного исчезновения клеток ганглиозного и зернистого слоя. Подобные изменения отмечают в нейронах передних рогов спинного мозга с демиелинпзациеи задних столбов, в подбугорной области. Со стороны поперечнополосатой мускулатуры вторичная атрофия. В печени очаговые некрозы и диффузная жировая инфильтрация с лимфоплазмоцитарной клеточной реакцией по портальному тракту.
В почках часто встречаются явления хронического пиелонефрита, в легких диффузные бронхоэктазы с абсцессами в сочетании с пневмосклерозом. Наблюдается атрофия яичек и яичников. В передней доле гипофиза дистрофические изменения ацидофильных клеток. Очень характерно сочетание AT с развитием злокачественных опухолей. Чаще встречаются злокачественные лимфомы, лимфогранулематоз, лейкозы, а также медуллобластомы, аденокарциномы, дизгерминомы, ретикулосаркомы.
Мы наблюдали злокачественную неходжкинскую лимфому забрюшинных лимфатических узлов с широкой генерализацией процесса, опухолевой инфильтрацией ткани тимуса у девочки 10 лет. Больные погибают от хронических заболеваний при явлениях общего истощения и выраженного инфантилизма или от злокачественных опухолей, чаще типа гемобластозов.
— Также рекомендуем «Иммунодефицит с ахондроплазией. Морфология иммунодефицита с ахондроплазией»
Оглавление темы «Врожденный иммунодефицит у плода и новорожденного»:
1. Акцидентальная инволюция тимуса. Этапы акцидентальной инволюции тимуса
2. Приобретенная атрофия тимуса. Первичные врожденные иммунодефицитные синдромы
3. Признаки врожденных иммунодефицитов. Дефицит клеточного иммунитета у новорожденного
4. Тяжелый комбинированный иммунодефицит. Морфология тимической алимфоплазии
5. Комбинированный иммунодефицит при ферментопатии. Синдром Незелофа или алимфоцитоз
6. Агенезия или гипоплазия тимуса — синдром Ди-Джорджи. Признаки агенезии тимуса
7. Иммунодефицитный синдром с атаксией-телеангиэктазией. Синдром Луи-Бар
8. Иммунодефицит с ахондроплазией. Морфология иммунодефицита с ахондроплазией
9. Агаммаглобулинемия, сцепленная с Х-хромосомой — синдром Брутона. Морфология синдрома Брутона
10. Дефицит иммуноглобулинов с повышением иммуноглобулинов М. Избирательный дефицит IgM и IgA
Источник