Синдром ларсена код по мкб

Наш генетический код настолько сложный, что практически любая серьезная поломка способна вызвать цепную реакцию и отразиться на человеке не с самой лучшей стороны. Ученые постоянно находят новые заболевания, но, по их собственным словам, на девяносто процентов геном остается неисследованным.
Описание
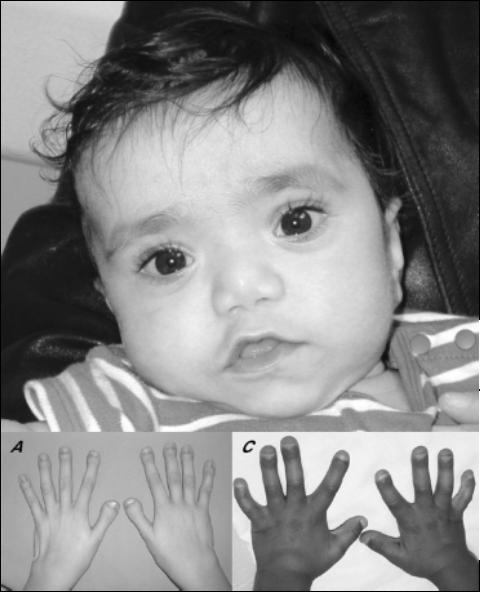
Синдром Ларсена (МКБ 10 – код М89) – это редко встречающееся генетическое заболевание, которое имеет широкий круг фенотипических проявлений. Наиболее характерными признаками считаются вывихи и подвывихи крупных суставов, наличие пороков развития костей лицевого черепа и проблемы с функциями конечностей. К второстепенным проявлениям относят сколиоз, косолапость, низкий рост и трудности с дыханием.
Синдром Ларсена вызывается точечными мутациями, которые могут произойти как спонтанно, так и быть переданными по наследству по аутосомно-доминантному типу. С изменениями в гене FLNB связывают целую группу заболеваний, проявляющихся нарушениями в скелетной системе. Конкретные проявления могут быть разными даже среди родственников.
Причины
Что же должно произойти в формирующемся организме, чтобы возник синдром Ларсена? Причины этого заболевания все еще скрываются в научном полумраке. Известно только, что для него характерным является аутосомно-доминантное наследование. То есть всего одной копии измененного гена будет достаточно, чтобы передать патологию своим детям, а может быть, даже внукам. Ген может быть получен от родителей (от обоих или от одного) либо являться результатом спонтанной мутации. Риск унаследовать данное заболевание — 50/50, независимо от пола ребенка и количества беременностей.
Измененный ген расположен в коротком плече третьей хромосомы. При желании исследователи могут точно указать место, где наследственная информация подверглась изменениям. В норме этот ген кодирует белок, известный в научных кругах как филамин В. Он играет значительную роль в развитии цитоскелета. Мутации приводят к тому, что белок перестает выполнять возложенные на него функции, и от этого страдают клетки организма.
У людей с данным синдромом возможен мозаицизм. То есть тяжесть и количество проявлений заболевания напрямую зависят от того, сколько клеток оказалось поражено. Некоторые люди могут даже не подозревать о том, что имеют дефект этого гена.
Эпидемиология

Синдром Ларсена с одинаковой частотой развивается как у мужчин, так и у женщин. По весьма приблизительным оценкам, данное заболевание встречается у одного новорожденного из ста тысяч. Это, по счастью, большая редкость. Оценки считаются недостоверными, потому что существуют определенные трудности в выявлении этого синдрома.
Впервые в медицинской литературе заболевание было описано в середине двадцатого века. Лорен Ларсен и соавторы нашли и зафиксировали шесть случаев проявления синдрома у детей.
Симптомы
Синдром Ларсена, как уже упоминалось выше, может по-разному проявляться даже между близкими родственниками. Наиболее характерными признаками заболевания считаются изменения костей лица. К ним относятся: широкая низкая переносица и широкий лоб, плоское лицо, наличие незаращения верхней губы или твердого неба. Кроме того, у детей имеются дислокации крупных суставов (бедренного, коленного, локтевого) и подвывихи плеча.
Пальцы у таких людей короткие, широкие, со слабыми разболтанными суставами. В запястьях могут присутствовать дополнительные кости, которые с возрастом сливаются и нарушают биомеханику движений. У некоторых людей наблюдается такой редкий феномен, как трахеомаляция (или размягчение хрящей трахеи).
Диагностика
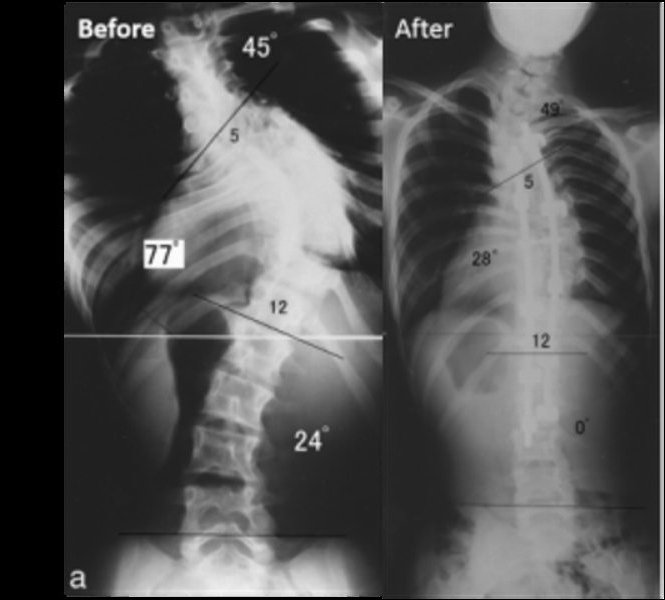
Диагноз «синдром Ларсена» ставится только после полного обследования больного, тщательного изучения его истории болезни и наличия характерных рентгенологических симптомов. Кроме того, полноценная радиографическая экспертиза может выявить и сопутствующие аномалии развития скелета, которые имеют косвенное отношение к заболеванию.
Ультразвуковая диагностика еще во внутриутробном периоде может выявить синдром Ларсена. Фото костных образований для хорошо подготовленного специалиста УЗИ может стать отправной точкой для поиска генетических аномалий плода. Так как с первого взгляда нельзя сказать, какая именно болезнь привела к патологии лицевого черепа и костей конечностей, будущей матери рекомендуют сделать амниоцентез и провести генетическую экспертизу для поиска мутации в третьей хромосоме.
Если заболевание подтверждается, но супруги приняли решение о сохранении беременности, то будущей матери рекомендуется провести процедуру кесарева сечения, чтобы не повредить кости ребенка в процессе прохождения его через таз женщины при естественных родах.
Лечение
Лечебные мероприятия направлены не на устранение заболевания, а на снижение клинических проявлений. Этим занимаются педиатры, ортопеды, специалисты по челюстно-лицевой хирургии и генетики. После оценки первоначального состояния ребенка и оценки всех рисков они могут приступить к коррекции имеющихся нарушений.
Самый щадящий вариант, которым лечится синдром Ларсена, – массаж. Он необходим для укрепления мышц и связок, удерживающих суставы, а также для улучшения поддержания спины и выпрямления позвоночника. Но до того как приступить к терапевтическим методам, потребуется ряд операций. Они необходимы для коррекции грубых деформаций скелета или уродств, стабилизации позвонков. При трахеомаляции требуется интубация, а затем и постановка дыхательной трубки (на постоянной основе), которая будет поддерживать проходимость верхних дыхательных путей.
Лечение этого заболевания – процесс длительный, который может растянуться на годы. С взрослением ребенка нагрузка на кости увеличивается, и ему снова может потребоваться физическая реабилитация, лечебная физкультура, а может быть, даже и хирургическое вмешательство.
Источник
Лабильная(ое)
— вазомоторная система T73.9
— кровяное давление R09.8
Лабиринт (ограниченный) (деструктивный) (диффузный) (внутреннего уха) (латентный) (гнойный) H83.0
— сифилитический A52.7† H94.8*
Лагофтальм (века) (нервный) H02.2
— с кератоконъюнктивитом H16.2
Ладонная(ый) — см. состояние
— фасция — см. состояние
Лайелла синдром L51.2
— вызванный лекарственным средством
— — при передозировке, неправильном назначении иди приеме лекарственного средства по ошибке T50.9
— — — уточненного лекарственного средства — см. Таблица лекарственных средств и химических веществ
— — при правильном введении соответствующего назначению лекарственного средства L51.2
Лайма болезнь A69.2
Лайтвуда-Олбрайта синдром N25.8
Лактация(и), лактационный (молочная железа) (послеродовая)
— мастит НКДР O91.2
— недостаточность (полная) O92.3
— — частичная O92.4
— не послеродовая N64.3
— период у матери (уход и/или обследование) Z39.1
— расстройство НКДР O92.7
— слабая (подавленная) O92.5
— чрезмерная O92.6
Лакунарный череп Q75.8
Ламберта-Итона синдром C80† G73.1*
Лангдона Дауна синдром (см. также Трисомия 21) Q90.9
Ландау-Клеффнера синдром F80.3
Ландри-Гийена-Барре синдром или паралич G61.0
Ландузи болезнь (иктерогеморрагический лептоспироз) A27.0
Ландузи-Дежерина дистрофия или плече-лопаточно-лицевая атрофия G71.0
Лансеро диабет E14.6
Larva migrans
— висцеральная НКДР B83.0
— кожная НКДР B76.9
— — вида Ancylostoma B76.0
Ларингизм (стридорный) J38.5
— врожденный Q31.4
— дифтерийный A36.2
Ларингит (острый) (отечный) (под собственно голосовым аппаратом) (гнойный) (язвенный) J04.0
— атрофический J37.0
— Венсана A69.1
— вызванный
— — внешним агентом (хронический) J37.0
— — Haemophilus influenzae O04.0
— гипертрофический J37.0
— гриппозный (см. также Грипп с респираторными проявлениями) J11.1
— дифтерийный A36.2
— катаральный J37.0
— обструктивный J05.0
— с (и)
— — гриппом (см. также Грипп с респираторными проявлениями) J11.1
— — трахеитом (острым) (см. также Ларинготрахеит) J04.2
— — — обструктивным J05.0
— — — хроническим J37.1
— сифилитический (поздний) A52.7† J99.8*
— — врожденный A50.5† J99.8*
— — — ранний A50.0† J99.8*
— спазматический J05.0
— — острый J04.0
— стрептококковый J04.0
— стридорный J05.0
— сухой J37.0
— туберкулезный A16.4
— — с бактериологическим и гистологическим подтверждением A15.5
— хронический J37.0
— — с трахеитом (хроническим) J37.1
Ларингоплегия J38.0
Ларингоптоз J38.7
Ларингоспазм J38.5
Ларингостеноз J38.6
Ларинготрахеит (острый) (инфекционный) J04.2
— атрофический J37.1
— Венсана A69.1
— вызванный
— — внешним агентом (хронический) J37.1
— — Haemophilus influenzae J04.2
— гипертрофический J37.1
— гриппозный (см. также Грипп с респираторными проявлениями) J11.1
— дифтерийный A36.2
— катаральный J37.1
— пахидермальный J38.7
— сифилитический (поздний) A52.7† J99.8*
— — врожденный A50.5† J99.8*
— — — ранний A50.0† O99.8*
— спазмотический J38.5
— — острый J05.0
— стрептококковый J04.2
— стридорный J38.5
— сухой J37.1
— туберкулезный A16.4
— хронический J37.1
Ларинготрахеобронхит (см. также Бронхит) J40
— острый (см. также Бронхит острый) J20.9
— хронический J42
Ларингофарингит (острый) J06.0
— хронический J37.0
Ларингофиссура J38.7
Ларингоцеле (врожденное) (вентрикулярное) Q31.3
Ларсена синдром Q74.8
Ларсена-Юханссона болезнь или остеохондроз M92.4
Ласса лихорадка A96.2
Латентный — см. состояние
Латероверсия
— матки (шейки) (постинфекционная) (послеродовая, старая) N85.4
— — врожденная Q51.8
— — при беременности и родах O34.5
— — — влияние на плод или новорожденного P03.8
— шейки матки — см. Латероверсия матки
Латерофлексия — см. Латероверсия
Латероцессия — см. Латероверсия
Латиризм T62.2
Лауна-Ганонга-Левина синдром I45.6
Лафора болезнь G40.3
Лаэннека цирроз K74.6
— алкогольный K70.3
Лгун патологический F60.2
Источник
Исключены:
- отдельные состояния, возникающие в перинатальном периоде (P00-P96)
- болезни височно-нижнечелюстного сустава (K07.6)
- некоторые инфекционные и паразитарные болезни (A00-B99)
- синдром сдавления (T79.6)
- осложнения беременности, родов и послеродового периода (O00-O99)
- врожденные аномалии, деформации и хромосомные нарушения (Q00-Q99)
- болезни эндокринной системы, расстройства питания и нарушения обмена веществ (E00-E90)
- травмы, отравления и некоторые другие последствия воздействия внешних причин (S00-T98)
- новообразования (C00-D48)
- симптомы, признаки и отклонения от нормы, выявленные при клинических и лабораторных исследованиях, не классифицированные в других рубриках (R00-R99)
Этот класс содержит следующие блоки:
- M00-M25 Артропатии
- M00-M03 Инфекционные артропатии
- M05-M14 Воспалительные полиартропатии
- M15-M19 Артрозы
- M20-M25 Другие поражения суставов
- M30-M36 Системные поражения соединительной ткани
- M40-M54 Дорсопатии
- M40-M43 Деформирующие дорсопатии
- M50-M54 Другие дорсопатии
- M60-M79 Болезни мягких тканей
- M60-M63 Поражения мышц
- M65-M68 Поражения синовиальных оболочек и сухожилий
- M70-M79 Другие поражения мягких тканей
- M80-M94 Остеопатии и хондропатии
- M80-M85 Нарушения плотности и структуры кости
- M86-M90 Другие остеопатии
- M91-M94 Хондропатии
- M95-M99 Другие нарушения костно-мышечной системы и соединительной ткани
Звездочкой отмечены следующие категории:
- M01* Прямое инфицирование сустава при инфекционных и паразитарных болезнях, классифицированных в других рубриках
- M03* Постинфекционные и реактивные артропатии при болезнях, классифицированных в других рубриках
- M07* Псориатические и энтеропатические артропатии
- M09* Ювенильный артрит при болезнях, классифицированных в других рубриках
- M14* Артропатии при других болезнях, классифицированных в других рубриках
- M36* Системные поражения соединительной ткани при болезнях, классифицированных в других рубриках
- M49* Спондилопатии ткани при болезнях, классифицированных в других рубриках
- M63* Поражения мышц при болезнях, классифицированных в других рубриках
- M68* Поражения синовиальных оболочек и сухожилий при болезнях, классифицированных в других рубриках
- M73* Поражения мягких тканей при болезнях, классифицированных в других рубриках
- M82* Остеопороз при болезнях, классифицированных в других рубриках
- M90* Остеопатии при болезнях, классифицированных в других рубриках
ЛОКАЛИЗАЦИЯ КОСТНО-МЫШЕЧНОГО ПОРАЖЕНИЯ
В классе XIII для обозначения локализации поражения введены дополнительные знаки, которые могут факультативно использоваться с соответствующими подрубриками. Поскольку место распространения или специальная адаптация могут варьироваться в количестве используемых цифровых характеристик, предполагается, что дополнительная подклассификация по локализации должна быть помещена в идентифицируемую отдельную позицию (например, в дополнительный блок). Различные подклассификации, используемые при уточнении повреждения колена, дорсопатиях или биомеханических нарушениях, не классифицированных в других рубриках, приведены в M23, в M40-M43 и в M99 соответственно
- 0 Множественная локализация
- 1 Плечевая область
- Ключица,
- акромиально-ключичный сустав,
- лопатка,
- плечевой сустав,
- грудино-ключичный сустав
- 2 Плечо
- Плечевая кость
- Локтевой сустав
- 3 Предплечье
- Лучевая кость
- Лучезапястный сустав,
- локтевая кость
- 4 Кисть
- Запястье,
- Суставы между этими костями
- пальцы,
- пясть
- 5 Тазовая область и бедро
- Ягодичная область
- Тазобедренный сустав,
- крестцо-подвздошный сустав
- бедренная кость,
- таз
- 6 Голень
- Малоберцовая кость,
- большеберцовая кость
- Коленный сустав
- 7 Голеностопный сустав и стопа
- Голеностопный сустав,
- Плюсна,
- предплюсна,
- другие суставы стопы пальцы стопы
- 8 Другие
- Голова, шея, ребра, череп, туловище, позвоночник
- 9 Локализация неуточненная
последние изменения: январь 2004
Следующие дополнительные пятые знаки, обозначающие локализацию поражения, даны для факультативного использования с соответствующими рубриками блока «Дорсопатии», исключая рубрики M50 и M51; см. также примечание в разделе M00-M99.
- 0 Множественные отделы позвоночника
- 1 Область затылка, первого и второго шейных позвонков
- 2 Область шеи
- 3 Шейно-грудной отдел
- 4 Грудной отдел
- 5 Пояснично-грудной отдел
- 6 Поясничный отдел
- 7 Пояснично-крестцовый отдел
- 8 Крестцовый и крестцово-копчиковый отдел
- 9 Неуточненная локализация
Источник
Исключены:
- лихорадка неясного происхождения (во время) (у):
- родов (O75.2)
- новорожденного (P81.9)
- лихорадка послеродового периода БДУ (O86.4)
Боль в области лица
Исключены:
- атипичная боль в области лица (G50.1)
- мигрень и другие синдромы головной боли (G43-G44)
- невралгия тройничного нерва (G50.0)
Включена: боль, которая не может быть отнесена к какому-либо определенному органу или части тела
Исключены:
- хронический болевой личностный синдром (F62.8)
- головная боль (R51)
- боль (в):
- животе (R10.-)
- спине (M54.9)
- молочной железе (N64.4)
- груди (R07.1-R07.4)
- ухе (H92.0)
- области таза (H57.1)
- суставе (M25.5)
- конечности (M79.6)
- поясничном отделе (M54.5)
- области таза и промежности (R10.2)
- психогенная (F45.4)
- плече (M25.5)
- позвоночнике (M54.-)
- горле (R07.0)
- языке (K14.6)
- зубная (K08.8)
- почечная колика (N23)
последние изменения: январь 2015
R53
Недомогание и утомляемость
Астения БДУ
Слабость:
- БДУ
- хроническая
Общее физическое истощение
Летаргия
Усталость
Исключены:
- слабость:
- врожденная (P96.9)
- старческая (R54)
- истощение и усталость (вследствие) (при):
- нервной демобилизации (F43.0)
- чрезмерного напряжения (T73.3)
- опасности (T73.2)
- теплового воздействия (T67.-)
- неврастении (F48.0)
- беременности (O26.8)
- старческой астении (R54)
- синдром усталости (F48.0)
- после перенесенного вирусного заболевания (G93.3)
последние изменения: январь 2012
Старческий возраст без упоминания о психозе
Старость без упоминания о психозе
Старческая:
- астения
- слабость
Исключен: старческий психоз (F03)
R55
Обморок [синкопе] и коллапс
Кратковременная потеря сознания и зрения
Потеря сознания
Исключены:
- нейроциркуляторная астения (F45.3)
- ортостатическая гипотензия (I95.1)
- неврогенная (G23.8)
- шок:
- БДУ (R57.9)
- кардиогенный (R57.0)
- осложняющий или сопровождающий:
- аборт, внематочную или молярную беременность (O00-O07, O08.3)
- роды и родоразрешение (O75.1)
- послеоперационный (T81.1)
- приступ Стокса-Адамса (I45.9)
- обморок:
- синокаротидный (G90.0)
- тепловой (T67.1)
- психогенный (F48.8)
- бессознательное состояние БДУ (R40.2)
последние изменения: январь 2016
Исключены: судороги и пароксизмальные приступы (при):
- диссоциативные (F44.5)
- эпилепсии (G40-G41)
- новорожденного (P90)
Исключены:
- шок (вызванный):
- анестезией (T88.2)
- анафилактический (вследствие):
- БДУ (T78.2)
- неблагоприятной реакции на пищевые продукты (T78.0)
- сывороточный (T80.5)
- осложняющий или сопровождающий аборт, внематочную или молярную беременность (O00-O07, O08.3)
- воздействием электрического тока (T75.4)
- в результате поражения молнией (T75.0)
- акушерский (O75.1)
- послеоперационный (T81.1)
- психический (F43.0)
- травматический (T79.4)
- синдром токсического шока (A48.3)
последние изменения: январь 2015
R58
Кровотечение, не классифицированное в других рубриках
Кровотечение БДУ
Включены: опухшие железы
Исключены: лимфаденит:
- БДУ (I88.9)
- острый (L04.-)
- хронический (I88.1)
- мезентериальный (острый) (хронический) (I88.0)
Исключена: задержка полового созревания (E30.0)
Исключены:
- булимия БДУ (F50.2)
- расстройства приема пищи неорганического происхождения (F50.-)
- недостаточность питания (E40-E46)
Исключены:
- синдром истощения как результат заболевания, вызванного ВИЧ (B22.2)
- злокачественная кахексия (C80.-)
- алиментарный маразм (E41)
последние изменения: январь 2010
Эта категория не должна использоваться в первичном кодировании. Категория предназначена для использования в множественном кодировании, чтобы определить данный синдром, возникший по любой причине. Первым должен быть присвоен код из другой главы, чтобы указать причину или основное заболевание.
добавлено: январь 2010
R69
Неизвестные и неуточненные причины заболевания
Болезненность БДУ
Недиагностированная болезнь без уточнения локализации или пораженной системы
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Крузона.
Синдром Крузона
Описание
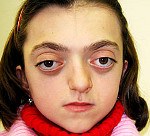
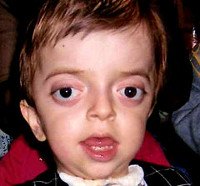
Синдром Крузона. Редкое генетическое заболевание, сопровождающееся прогрессирующими деформациями лицевой и мозговой части черепа и краниосиностозом с развитием сопутствующих нарушений. Симптомами этого состояния являются изменение формы головы (брахицефалия, скафоцефалия, тригоноцефалия), крючковидный нос, гипоплазия средней трети лица, нарушения зрения и слуха. Диагностика синдрома Крузона осуществляется на основании внешних проявлений заболевания, рентгенологических данных, а также молекулярно-генетических анализов. Специфического лечения этой патологии не существует, используются паллиативные и симптоматические мероприятия, в том числе хирургического характера.
Дополнительные факты
Синдром Крузона (краниофасциальный дизостоз 1 типа) – генетическое заболевание, характеризующееся нарушением процессов окостенения и развития элементов скелета лицевого и мозгового черепа. Впервые это состояние было описано в 1912 году французским педиатром О. Крузоном, с тех пор синдром носит его имя. Механизм наследования синдрома Крузона – аутосомно-доминантный, однако заболевание часто обусловлено спонтанными мутациями. Патология встречается достаточно редко – примерно 1,6 случаев на 100 000 новорожденных, при этом данным синдромом обусловлено почти 5% от всех пороков развития, сопровождающихся черепным дизостозом. Долгое время считалось, что это состояние имеет две разновидности – обычную и сопровождающуюся кожными нарушениями (гиперкератозом, акантозом), но с учетом современных данных специалисты в области генетики установили, что синдром Крузона с черным акантозом (CAN) является отдельным наследственным заболеванием. В то же время, патогенез его развития аналогичен классической форме заболевания, именно этим объясняется значительная схожесть симптомов. Состояние с одинаковой вероятностью поражает как мальчиков, так и девочек.
Синдром Крузона
Причины
Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме – он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов, что снижает его стабильность – в нем часто развиваются дефекты, приводящие к многочисленным генетическим заболеваниям, в основном поражающим элементы скелета. Так, помимо синдрома Крузона, мутации FGFR2 могут быть причиной синдромов Апера, Сетре-Чотзена, Бира-Стивенсона, синдрома Пфайффера и многих других патологий. Генетические исследования показали, что краниофасциальный дизостоз 1-го типа способны вызывать более 35 мутаций вышеуказанного гена, в основном они локализованы в области 7 и 9 экзонов.
Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка. Изменение конформации рецептора к фактору роста фибробластов 2 нарушает межклеточные взаимодействия в соединительных тканях черепа, главным образом костной и хрящевой. Это приводит сначала к накоплению фибробластов в области межкостных швов, а потом к активизации процессов окостенения, что и является причиной ведущего проявления синдрома Крузона – черепного синостоза. Некоторые исследователи полагают, что данные генетические дефекты влияют также на эмбриональное развитие структур первой жаберной дуги – к ним относят челюсти и отчасти элементы средней трети лица. Именно этим объясняется гипоплазия челюстей, особенно нижней, при синдроме Крузона.
Причины синдрома Крузона с черным акантозом несколько иные – он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями – Ala391Glu Это тоже миссенс-мутация, изменяющая структуру белка-рецептора. Патогенез заболевания практически не отличается от классического варианта. Изменения лица и черепа при синдроме Крузона с черным акантозом аналогичны предыдущему типу, однако к ним присоединяются гиперкератоз различных участков кожи и акантоз, нередко наблюдаются многочисленные родинки.
Симптомы
Проявления синдрома Крузона можно заметить уже при рождении ребенка, однако наиболее выраженными они становятся на протяжении первых 3-4 лет жизни. Самым характерным симптомом заболевания является краниосиностоз, который может развиваться на венечном или стреловидном (намного реже) шве, прочно соединяя кости и останавливая нормальный рост головы. Сразу после рождения первые признаки синостоза могут быть стертыми, но всегда наблюдается гипертелоризм, прогнатия нижней челюсти, изменение формы носа по типу «клюва попугая», незначительный экзофтальм из-за уменьшенного размера глазниц, низкое расположение наружного слухового прохода. Иногда при синдроме Крузона выявляется синдактилия пальцев, в этом случае необходимо производить дифференциальную диагностику с синдромом Апера. У некоторых больных обнаруживается атрезия хоан, затрудняющая дыхание, а также гидроцефалия, еще больше осложняющая течение заболевания за счет резкого возрастания внутричерепного давления.
Особенностью синдрома Крузона является неминуемое прогрессирование заболевания, особенно в отношении формы черепа. Из-за образования прочного синостоза и продолжающегося роста размеров головного мозга форма головы изменяется, возникает брахицефалия или «башенный череп» – в зависимости от того, по какому шву произошло срастание. При синдроме Крузона в области сросшихся костей черепа также могут образовываться экзостозы. Такая деформация приводит и к поражению органов зрения – сначала возникает расходящееся косоглазие, затем экзофтальм сильно прогрессирует вплоть до выпадения глазных яблок из орбиты. Нередко синдром Крузона сопровождается расстройствами слуха из-за нарушения структуры пирамиды височной кости – ее полости уменьшены в размерах, некоторые из них могут отсутствовать, нередко это приводит к полной глухоте. Наблюдаются изменения и со стороны нервной системы, обнаруживаются нарастающие признаки умственной отсталости (при отсутствии паллиативных мероприятий), симптомы повышения внутричерепного давления (головные боли, рвота), судорожные припадки.
Рвота. Судороги.
Диагностика
Выявление синдрома Крузона возможно на этапе пренатального развития, сразу после рождения или в первые годы жизни больного. Для этого применяются рентгенологические методики, общий осмотр, молекулярно-генетические анализы. Вспомогательную роль в диагностике синдрома Крузона играют такие методы, как офтальмологический осмотр, исследование слуха, оценка интеллектуального и психического развития. При осмотре маленьких детей определяются низко посаженные уши, гипоплазия средней трети лица, экзофтальм. У больных синдромом Крузона старшего возраста к этим проявлениям присоединяются расходящееся косоглазие, ослабление слуха вплоть до полной глухоты, изменение формы черепа. На рентгенографии черепа регистрируется синостоз в области венечного, стреловидного или лямбдовидного швов, возможно обнаружение экзостозов и уплощенной формы глазниц.
Томография пирамиды височной кости при синдроме Крузона выявляет нарушение формирования наружного слухового прохода (атрезия или стеноз) и других полостей, иногда наблюдается отсутствие барабанной полости. Турецкое седло несколько расширено, могут образовываться добавочные мелкие околоносовые синусы. Молекулярно-генетическая диагностика синдрома Крузона производится врачом-генетиком и при классической форме заболевания сводится к автоматическому секвенированию 7 и 9 экзонов гена FGFR2 с целью выявления мутаций. При наличии кожных проявлений (гиперкератоза, бородавках, множественных родинках) имеет смысл производить поиск мутации Ala391Glu в гене FGFR3. Для обеих форм синдрома Крузона возможна пренатальная генетическая диагностика, ультразвуковые методики при этом, как правило, малоэффективны.
Лечение
Какого-либо специфического лечения синдрома Крузона на сегодняшний момент не существует, применяют только паллиативные мероприятия. К ним относят хирургические вмешательства по ремоделированию формы черепа и устранению синостозов – такие процедуры необходимо начинать как можно раньше и в дальнейшем производить еще несколько раз по мере роста головы. Это снижает уровень внутричерепного давления, что положительно сказывается на умственном развитии больных синдромом Крузона и уменьшает вероятность появления неврологических нарушений. Также с помощью хирургических методик создают искусственный блефарофимоз для снижения степени экзофтальма и предотвращения вывиха глазного яблока. При атрезии хоан производится их расширение оперативным путем для облегчения дыхания. Описаны техники радикальных комплексных операций, направленных на устранение большинства лицевых нарушений при синдроме Крузона. В случае развития кожных изменений для снижения их выраженности рекомендуется наружное применение средств на основе ретиноидов, иногда назначают кортикостероиды.
Прогноз синдрома Крузона, как правило, неопределенный, многие специалисты оценивают его как неблагоприятный. Это связано с тем, что даже при проведении всех симптоматических и паллиативных мероприятий у больных все равно нарастает расходящееся косоглазие, практически всегда со временем развивается глухота, гипоплазия средней трети лица становится более выраженной с возрастом. Тем не менее, многие больные при соответствующем лечении и уходе могут доживать до преклонного возраста. По причине сильного нарушения зрения и слуха практически всегда происходит инвалидизация пациентов, причиной инвалидности также может стать умственная отсталость. Профилактика синдрома Крузона не разработана, возможно лишь пренатальное определение патологии молекулярно-генетическими методами.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник