Синдром крузона код по мкб 10

Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Крузона.
Синдром Крузона
Описание
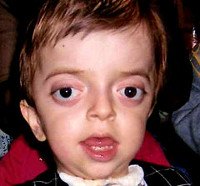
Синдром Крузона. Редкое генетическое заболевание, сопровождающееся прогрессирующими деформациями лицевой и мозговой части черепа и краниосиностозом с развитием сопутствующих нарушений. Симптомами этого состояния являются изменение формы головы (брахицефалия, скафоцефалия, тригоноцефалия), крючковидный нос, гипоплазия средней трети лица, нарушения зрения и слуха. Диагностика синдрома Крузона осуществляется на основании внешних проявлений заболевания, рентгенологических данных, а также молекулярно-генетических анализов. Специфического лечения этой патологии не существует, используются паллиативные и симптоматические мероприятия, в том числе хирургического характера.
Дополнительные факты
Синдром Крузона (краниофасциальный дизостоз 1 типа) – генетическое заболевание, характеризующееся нарушением процессов окостенения и развития элементов скелета лицевого и мозгового черепа. Впервые это состояние было описано в 1912 году французским педиатром О. Крузоном, с тех пор синдром носит его имя. Механизм наследования синдрома Крузона – аутосомно-доминантный, однако заболевание часто обусловлено спонтанными мутациями. Патология встречается достаточно редко – примерно 1,6 случаев на 100 000 новорожденных, при этом данным синдромом обусловлено почти 5% от всех пороков развития, сопровождающихся черепным дизостозом. Долгое время считалось, что это состояние имеет две разновидности – обычную и сопровождающуюся кожными нарушениями (гиперкератозом, акантозом), но с учетом современных данных специалисты в области генетики установили, что синдром Крузона с черным акантозом (CAN) является отдельным наследственным заболеванием. В то же время, патогенез его развития аналогичен классической форме заболевания, именно этим объясняется значительная схожесть симптомов. Состояние с одинаковой вероятностью поражает как мальчиков, так и девочек.
Синдром Крузона
Причины
Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме – он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов, что снижает его стабильность – в нем часто развиваются дефекты, приводящие к многочисленным генетическим заболеваниям, в основном поражающим элементы скелета. Так, помимо синдрома Крузона, мутации FGFR2 могут быть причиной синдромов Апера, Сетре-Чотзена, Бира-Стивенсона, синдрома Пфайффера и многих других патологий. Генетические исследования показали, что краниофасциальный дизостоз 1-го типа способны вызывать более 35 мутаций вышеуказанного гена, в основном они локализованы в области 7 и 9 экзонов.
Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка. Изменение конформации рецептора к фактору роста фибробластов 2 нарушает межклеточные взаимодействия в соединительных тканях черепа, главным образом костной и хрящевой. Это приводит сначала к накоплению фибробластов в области межкостных швов, а потом к активизации процессов окостенения, что и является причиной ведущего проявления синдрома Крузона – черепного синостоза. Некоторые исследователи полагают, что данные генетические дефекты влияют также на эмбриональное развитие структур первой жаберной дуги – к ним относят челюсти и отчасти элементы средней трети лица. Именно этим объясняется гипоплазия челюстей, особенно нижней, при синдроме Крузона.
Причины синдрома Крузона с черным акантозом несколько иные – он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями – Ala391Glu Это тоже миссенс-мутация, изменяющая структуру белка-рецептора. Патогенез заболевания практически не отличается от классического варианта. Изменения лица и черепа при синдроме Крузона с черным акантозом аналогичны предыдущему типу, однако к ним присоединяются гиперкератоз различных участков кожи и акантоз, нередко наблюдаются многочисленные родинки.
Симптомы
Проявления синдрома Крузона можно заметить уже при рождении ребенка, однако наиболее выраженными они становятся на протяжении первых 3-4 лет жизни. Самым характерным симптомом заболевания является краниосиностоз, который может развиваться на венечном или стреловидном (намного реже) шве, прочно соединяя кости и останавливая нормальный рост головы. Сразу после рождения первые признаки синостоза могут быть стертыми, но всегда наблюдается гипертелоризм, прогнатия нижней челюсти, изменение формы носа по типу «клюва попугая», незначительный экзофтальм из-за уменьшенного размера глазниц, низкое расположение наружного слухового прохода. Иногда при синдроме Крузона выявляется синдактилия пальцев, в этом случае необходимо производить дифференциальную диагностику с синдромом Апера. У некоторых больных обнаруживается атрезия хоан, затрудняющая дыхание, а также гидроцефалия, еще больше осложняющая течение заболевания за счет резкого возрастания внутричерепного давления.
Особенностью синдрома Крузона является неминуемое прогрессирование заболевания, особенно в отношении формы черепа. Из-за образования прочного синостоза и продолжающегося роста размеров головного мозга форма головы изменяется, возникает брахицефалия или «башенный череп» – в зависимости от того, по какому шву произошло срастание. При синдроме Крузона в области сросшихся костей черепа также могут образовываться экзостозы. Такая деформация приводит и к поражению органов зрения – сначала возникает расходящееся косоглазие, затем экзофтальм сильно прогрессирует вплоть до выпадения глазных яблок из орбиты. Нередко синдром Крузона сопровождается расстройствами слуха из-за нарушения структуры пирамиды височной кости – ее полости уменьшены в размерах, некоторые из них могут отсутствовать, нередко это приводит к полной глухоте. Наблюдаются изменения и со стороны нервной системы, обнаруживаются нарастающие признаки умственной отсталости (при отсутствии паллиативных мероприятий), симптомы повышения внутричерепного давления (головные боли, рвота), судорожные припадки.
Рвота. Судороги.
Диагностика
Выявление синдрома Крузона возможно на этапе пренатального развития, сразу после рождения или в первые годы жизни больного. Для этого применяются рентгенологические методики, общий осмотр, молекулярно-генетические анализы. Вспомогательную роль в диагностике синдрома Крузона играют такие методы, как офтальмологический осмотр, исследование слуха, оценка интеллектуального и психического развития. При осмотре маленьких детей определяются низко посаженные уши, гипоплазия средней трети лица, экзофтальм. У больных синдромом Крузона старшего возраста к этим проявлениям присоединяются расходящееся косоглазие, ослабление слуха вплоть до полной глухоты, изменение формы черепа. На рентгенографии черепа регистрируется синостоз в области венечного, стреловидного или лямбдовидного швов, возможно обнаружение экзостозов и уплощенной формы глазниц.
Томография пирамиды височной кости при синдроме Крузона выявляет нарушение формирования наружного слухового прохода (атрезия или стеноз) и других полостей, иногда наблюдается отсутствие барабанной полости. Турецкое седло несколько расширено, могут образовываться добавочные мелкие околоносовые синусы. Молекулярно-генетическая диагностика синдрома Крузона производится врачом-генетиком и при классической форме заболевания сводится к автоматическому секвенированию 7 и 9 экзонов гена FGFR2 с целью выявления мутаций. При наличии кожных проявлений (гиперкератоза, бородавках, множественных родинках) имеет смысл производить поиск мутации Ala391Glu в гене FGFR3. Для обеих форм синдрома Крузона возможна пренатальная генетическая диагностика, ультразвуковые методики при этом, как правило, малоэффективны.
Лечение
Какого-либо специфического лечения синдрома Крузона на сегодняшний момент не существует, применяют только паллиативные мероприятия. К ним относят хирургические вмешательства по ремоделированию формы черепа и устранению синостозов – такие процедуры необходимо начинать как можно раньше и в дальнейшем производить еще несколько раз по мере роста головы. Это снижает уровень внутричерепного давления, что положительно сказывается на умственном развитии больных синдромом Крузона и уменьшает вероятность появления неврологических нарушений. Также с помощью хирургических методик создают искусственный блефарофимоз для снижения степени экзофтальма и предотвращения вывиха глазного яблока. При атрезии хоан производится их расширение оперативным путем для облегчения дыхания. Описаны техники радикальных комплексных операций, направленных на устранение большинства лицевых нарушений при синдроме Крузона. В случае развития кожных изменений для снижения их выраженности рекомендуется наружное применение средств на основе ретиноидов, иногда назначают кортикостероиды.
Прогноз синдрома Крузона, как правило, неопределенный, многие специалисты оценивают его как неблагоприятный. Это связано с тем, что даже при проведении всех симптоматических и паллиативных мероприятий у больных все равно нарастает расходящееся косоглазие, практически всегда со временем развивается глухота, гипоплазия средней трети лица становится более выраженной с возрастом. Тем не менее, многие больные при соответствующем лечении и уходе могут доживать до преклонного возраста. По причине сильного нарушения зрения и слуха практически всегда происходит инвалидизация пациентов, причиной инвалидности также может стать умственная отсталость. Профилактика синдрома Крузона не разработана, возможно лишь пренатальное определение патологии молекулярно-генетическими методами.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: Q75.1
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q65-Q79 Врожденные аномалии пороки развития и деформации костно-мышечной системы / Q75 Другие врожденные аномалии (пороки развития) костей черепа и лица
Определение и общие сведения[править]
Синдром Крузона
Синонимы: черепно-лицевой дизостоз Крузона, «попугайная» болезнь,
Синдром Крузона — краниостеноз, обусловленный сочетанием недоразвития костей черепа и преждевременным зарастанием черепных швов, что проявляется изменением формы мозгового и лицевого черепа.
Синдром Крузона описал в 1912 г. французский врач O. Crouzon (1874-1938).
Частота распространенности синдрома Крузона в США оценивается как 1 случай на 60 000 живорожденных (приблизительно 16,6 случая на 1 000 000 жителей). По оценкам, распространенность в общей популяции Европы в 0,9 / 100000.
Синдром Крузона составляет около 4,8% от всех случаев краниосиностозов.
Тип наследования синдрома аутосомно-доминантный, с переменной пенетрантностью.
Этиология и патогенез[править]
Синдром Крузона вызывается мутациями рецептора фактора роста фибробластов гена FGFR2 (10q25.3-В26), 80% мутаций расположены в иммуноглобулино-подобном домене III (IgIII домен) внеклеточной области и 20% мутаций расположены в IgI-IgII доменах трансмембранной и тирозинкиназной зоны генома.
Отдельная форма заболевания черным акантозом рассматривается ниже.
Мутации гена ERF (19q13.2), кодирующего ETS2 репрессор, что приводит к стимуляции аностеогенеза связаны с возникновением Крузона-подобного синдрома
Клинические проявления[править]
При этом характерны гипертелоризм, экзофтальм, страбизм, своеобразная крючковатая форма носа, напоминающего клюв орла или попугая, возможны недоразвитие нижней челюсти, нарушение прикуса: нижние зубы впереди верхних (прогнатия).
Гидроцефалия, опущение миндалин мозжечка и аномалии венозного оттока яремной вены также часто наблюдается при болезни Крузона и могут создавать терапевтические проблемы. Две трети пациентов с болезнью Крузона имеют внутричерепную гипертензию, что может привести к слепоте.
Краниофациальный дизостоз: Диагностика[править]
На глазном дне нередко отмечаются признаки застоя, который может смениться вторичной атрофией дисков зрительных нервов, сопровождающейся нарушением зрения.
Дифференциальный диагноз[править]
Краниофациальный дизостоз: Лечение[править]
Хирургические вмешательства направлены на предотвращение церебральных, офтальмологические или респираторных осложнений и коррекции аномалий черепно-лицевых размеров и формы.
Профилактика[править]
Прочее[править]
Синдром Крузона — черный акантоз
Синдром Крузона с черным акантозом является очень редкой, клинически гетерогенной формой фациокраниостеноза с Крузона-подобными проявлениями в виде преждевременного синостоза черепных швов (синдром Крузона) и черного акантоза.
По оценкам распространенность порядка 1/1000000 новорожденных. Менее 70 случаев заболевания были описаны в медицинской литературе на настощий момент. Соотношение полов — 2,4:1 с превалированием девочек.
Этиология и патогенез
Синдром Крузона с черным акантозом вызван специфической мутацией p.Ala391Glu рецептора 3 фактора роста фибробластов гена FGFR3 (4p16.3), участвующего в регуляции клеточной пролиферации, дифференцировки и апоптоза. Возникновение черного акантоза связано недостаточной стимуляцией рецепторов различных факторов роста фибробластов.
Клинические проявления
Все пациенты имеют врожденные аномалии черепно-лицевых костей в соответствии с картиной классического синдрома Крузона, включая краниосиностоз, гипоплазию средней части лица, неглубокие орбиты с экзофтальмом, скошенные книзу глазные щели и гипертелоризм, верхнечелюстную гипоплазию с выпуклым носом и угловатые уши, смещенные кзади. Синостоз обычно затрагивает корональные швы черепа. Также сообщалось о случаях клеверообразного (трилистник) черепа. Пациенты также обнаруживают бархатистую гиперпигментацию кожи (черный акантоз) в течение первого десятилетия жизни, в основном в складках на шее, подмышечных впадинах, веках, периоральной, паховой и перианальной областей. Поражения кожи иногда широко представлены на теле и развиваются рано, по сравнению с классическим черным акантозом.
Аномалии краниовертебрального сочленения и позвоночника, такие как незначительные отклонения расстояние между позвонками (interpediculate distance) дистального позвоночника являются непостоянным симптомом синдрома Крузона — черный акантоз. Часто отмечается атрезия или стеноз хоан (41%). Другие часто сообщаемые симптомы включают гидроцефалию (43%), аномалии формирования нёба и прикуса, цементомы челюсти (34%) и меланоцитный невус (25%). Иногда наблюдаются поражение почек. Некоторые из вышеперечисленных симптомов встречаются редко у пациентов с классическим синдромом Крузона. Снижение интеллекта, потеря слуха и задержка речи являются редкостью для синдром Крузона с черным акантозом. Тяжесть поражений одинакова у малтчков и девочек.
Генетическая консультация
В большинстве случаев заболевание имеет спорадический характер, часто связано с поздним возрастом родителей, хотя семейные случаи, тем не менее есть данные о семейных случаях с аутосомно-доминантным типом наследования.
Источники (ссылки)[править]
Общая неврология [Электронный ресурс] / А. С. Никифоров, Е. И. Гусев. — 2-е изд., испр. и доп. — М. : ГЭОТАР-Медиа, 2015. — https://www.rosmedlib.ru/book/ISBN9785970433850.html
Черепно-лицевая хирургия в формате 3D [Электронный ресурс] : атлас / Бельченко В.А., Притыко А.Г., Климчук А.В., Филлипов В.В. — М. : ГЭОТАР-Медиа, 2010.
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Синдром Крузона – редкое генетическое заболевание, сопровождающееся прогрессирующими деформациями лицевой и мозговой части черепа и краниосиностозом с развитием сопутствующих нарушений. Симптомами этого состояния являются изменение формы головы (брахицефалия, скафоцефалия, тригоноцефалия), крючковидный нос, гипоплазия средней трети лица, нарушения зрения и слуха. Диагностика синдрома Крузона осуществляется на основании внешних проявлений заболевания, рентгенологических данных, а также молекулярно-генетических анализов. Специфического лечения этой патологии не существует, используются паллиативные и симптоматические мероприятия, в том числе хирургического характера.
Причины заболевания
Синдром Крузона — наследственная патология, возникающая вследствие мутационного процесса. Передается недуг по аутосомно-доминантному принципу от родителей детям. Когда в одной родительской хромосоме присутствует мутировавший ген, дети не всегда наследуют заболевание. В таких семьях риск рождения ребенка с синдромом Крузона составляет 50%. Причем новорожденные дети могут даже не быть носителями дефекта. Тщательное обследование на этапе планирования беременности позволяет повысить шанс рождения здорового малыша.

Данный недуг развивается при мутации гена, расположенной в X хромосоме. Этот ген является нестабильным из-за значительных размеров и большого количества экзонов. Его мутации приводят к развитию наследственных патологий скелета человека. Происходит нарушение структуры соединительной ткани, костей и хрящей. В области межкостных швов скапливаются фибробласты, синтезирующие межклеточное вещество, продуцирующие белки — коллаген и эластин и активизирующие процессы окостенения. Это основной этиопатогенетический фактор синдрома Крузона.
К факторам, способствующим развитию патологии, относятся: наличие патологии в семейном анамнезе, возраст отца старше 60 лет в период зачатия ребенка.
Известны случаи ненаследственного характера синдрома Крузона, развивающегося на фоне спонтанной мутации в гене. Чаще всего такие нарушения встречаются по отцовской линии.
Симптомы
При синдроме Крузона у ребенка отмечается нарушения не только костной системы черепа, но и патологии органов, отвечающих за зрение и слух. Таким образом, клиническая картина аномалии весьма яркая и обширная.
| Костная система | Органы слуха | Органы зрения |
|---|---|---|
| • деформация черепной коробки; • деформация средней части лица; • изменение формы носа; • изменение челюстно-лицевой системы, при которых челюсти не могут полностью смыкаться; • нижняя губа ребенка недоразвита, расположена достаточно низко; • язык ребенка периодически выступает за пределы зубного ряда; • в некоторых случаях на черепе можно нащупать специфические гладкие швы. | • нарушение слуха, глухота различной степени тяжести; • деформация внутреннего слухового прохода; • снижение слуховой проводимости костей черепа; • изменение строения внешнего слухового канала. | • пучеглазость, выпячивание глаз, при котором строение глазного яблока не меняется; • колебания глазных яблок, носящие непроизвольный характер; • косоглазие, при котором глаза как бы смотрят в разные стороны; • расстояние межу внутренними уголками глаз и зрачками увеличено; • неправильное расположение хрусталика глаза или зрачка, когда он расположен не по центру, как это должно быть; • деформация радужки (часть ее может отсутствовать) и роговицы (увеличение ее размеров). |
Осложнения и последствия
Синдром Крузона не может пройти бесследно: как правило, у ребенка остаются различные последствия и осложнения:
- гидроцефалия;
- ухудшение зрения, вплоть до потери (вследствие продолжительного сдавливания зрительного нерва в нем происходят необратимые изменения);
- истончение и язвенное повреждение роговицы (из-за чрезмерной выпуклости глазных яблок становится невозможным полное смыкание век, в результате чего роговица частично высыхает и покрывается язвами);
- умственная отсталость;
- трудности с адаптацией в обществе (умственная неполноценность и неприятные внешние проявления синдрома значительно затрудняют взаимодействие пациента с социумом).
Ещё одним осложнением синдрома может стать аномалия Арнольда-Киари – это перемещение миндалин мозжечка сквозь большое затылочное отверстие к шейным позвонкам.
Лечение синдрома
Как таковое, лечение синдрома Крузона невозможно. Возможно лишь проведение функциональной и/или косметической хирургической коррекции, заключающейся в частичном удалении преждевременно синостозированных швов, а также предотвращение вывиха глазного яблока путем хирургического создания блефарофимоза. Французский хирург P. Tessier детально описал радикальную хирургическую операцию, которая сможет максимально исправить деформации лица при синдроме Крузона. Прогноз синдрома Крузона крайне неутешителен. С возрастом отмечается содружественное, альтернирующее или расходящееся косоглазие. Бинокулярное зрение становится невозможным. Зачастую отмечается атрофия зрительного нерва, приводящая к абсолютной потере зрения. В некоторых случаях возможно полное выпадение одного/обоих глазных яблок, которое происходит вследствие прогрессирующего уплощения глазниц. В области брегмы наблюдается образование экзостозов, вследствие преждевременного синостозирования швов. С возрастом, при нормальном развитии нижней челюсти, все более выраженной становится гипоплазия средней части лица.
Прогноз и профилактика заболевания
Прогноз синдрома Крузона, как правило, неопределенный, многие специалисты оценивают его как неблагоприятный. Это связано с тем, что даже при проведении всех симптоматических и паллиативных мероприятий у больных все равно нарастает расходящееся косоглазие, практически всегда со временем развивается глухота, гипоплазия средней трети лица становится более выраженной с возрастом. Тем не менее, многие больные при соответствующем лечении и уходе могут доживать до преклонного возраста. По причине сильного нарушения зрения и слуха практически всегда происходит инвалидизация пациентов, причиной инвалидности также может стать умственная отсталость. Профилактика синдрома Крузона не разработана, возможно лишь пренатальное определение патологии молекулярно-генетическими методами.
Загрузка…
Источник