Синдром корнелии де ланга фото

Многие из школьных уроков биологии помнят, что наследственная информация, передающаяся от отца с матерью их детям, заключается в геноме человека, состоящем из 23 пар хромосом. В них содержатся около 28 тысяч генов, каждый из которых играет свою важнейшую роль в формировании человеческого организма. Мутационные изменения только одного из них могут спровоцировать синдром Корнелии де Ланге, очень неприятное, а в большинстве случаев и достаточно тяжелое заболевание, нередко приводящее к летальному исходу. Многие авторы утверждают, что у страдающих этим синдромом нет шансов. И тем не менее не стоит отчаиваться, потому что современные наука и медицина творят настоящие чудеса.
Этимология названия
Синдром Корнелии де Ланге был так назван в честь подробно его описавшего детского врача по имени Корнелия де Ланге, в 30-х годах ХХ века жившего и работавшего в Голландии. Она в своей практике наблюдала 5 подобных случаев, в последний раз сразу двух девочек, не являвшихся родственницами. В 1933 году Корнелия сделала подробное описание и заключение своих наблюдений. Но гораздо раньше (в 1916 году) это же заболевание диагностировал и подробно описывал немецкий врач В. Брахман, поэтому часто используют название синдром Брахмана — Ланге. Кроме того, встречается название амстердамский синдром, так как в этом городе зафиксировано сразу трое детей, у которых обнаружилась такая патология. Все три наименования – это одно и то же заболевание. Оно встречается на всех континентах, у людей всех рас и этнических групп, с одинаковой частотой как у мальчиков, так и у девочек. За почти сто лет, прошедших с момента первого описания, подробно изучено около 400 случаев данной патологии.

Аномалии головы и кожи
Синдром Корнелии де Ланге можно заподозрить с первых минут жизни младенца. Первичные симптомы:
1. Малый вес новорожденного (примерно 2/3 от нормы).
2. Аномалии черепа:
— микроцефалия (череп примерно на 10 % меньше нормы (у 98 % больных);
— брахицефалия (увеличение поперечного размера (ширины) черепа по сравнению с его продольными размерами);
— уменьшение мозговой части головы.
Больше чем у половины новорожденных наблюдается повышенная волосистость спины, а иногда и всего тела. Кожа у примерно 2/3 младенцев имеет синюшный рисунок с хорошо просматриваемыми сосудами (мраморность кожи), но этот симптом не является определяющим в диагностике данного заболевания.
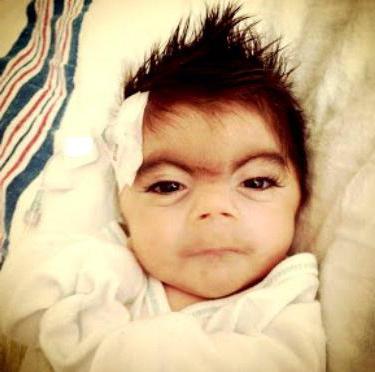
Аномалии лица
Дефекты на лице новорожденного являются наиболее яркими критериями, чтобы диагностировать синдром Корнелии Де Ланге. Фото малыша выше наглядно это показывает. К отклонениям от общепринятых норм относятся:
— четко очерченные, как нарисованные брови, сходящиеся на переносице (99 % случаев);
— красивые, длинные ресницы, часто загнутые назад (99 %);
— маленький носик, на котором ноздри выпячиваются вперед (88 %);
— широкая и запавшая переносица (88 %);
— рот с опущенными уголками (94%);
— нестандартно большое расстояние между носом и губами;
— низко расположенные уши;
— недоразвитие (гипоплазия) нижней челюсти (84 %);
— слишком низко расположена на лбу и/или затылке граница волосяного покрова (94 %).
Вышеперечисленные отклонения на лице новорожденного могут присутствовать все, а могут только некоторые.
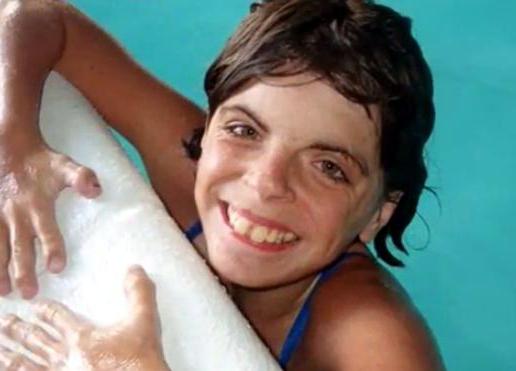
Аномалии внутренних органов
Гипоплазия (недоразвитие) и патологические нарушения в формировании внутренних органов представляют наибольшую опасность для жизни детей, у которых выявлен синдром Корнелии де Ланге. Диагностика включает МРТ, рентген, УЗИ, риноскопию, цитогенетические анализы и другие современные методы.
При данном заболевании может наблюдаться:
— атрезия хоан, а проще говоря, непроходимость полости носа (эта патология бывает совершенно не связана с синдромом де Ланге и легко диагностируется по тому, как младенец дышит либо с помощью зонда);
— дефекты в строении сердца (пороки сосудов, клапанов, перегородок);
— пороки ЖКТ (слепая кишка подвижна, некоторые другие);
— пороки мочеполовой системы (примерно у 50 % больных);
— кисты на почках, гидронефроз;
— патологии в мозговой ткани (дисплазия извилин, аплазия мозолистого тела и другие);
— слишком высокое либо с расщелиной нёбо;
— крипторхизм.
Опять-таки необязательно, чтобы у одного больного ребенка присутствовали все вышеперечисленные дефекты внутренних органов.
Аномалии костно-мышечного аппарата
У новорожденных могут быть дефекты конечностей, позвоночника, грудной клетки, по которым также диагностируется синдром Корнелии Де Ланге (фото людей с этим заболеванием представлены в статье).
Некоторые дефекты видны сразу. Это:
— отсутствие одного или нескольких пальцев на руках;
— сросшиеся пальцы (чаще встречается на ногах);
— деформированные позвоночник и/или грудная клетка.
С взрослением ребенка становятся довольно значительными следующие отклонения:
— отставание в росте (в некоторых случаях нанизм);
— недоразвитие, укорочение конечностей;
— маленькие кисти рук и стоп;
— слишком короткая шея;
— ограничение в способности локтевых суставов сгибаться-разгибаться (контрактура).
Неврологические отклонения и патологии органов чувств
К сожалению, есть множество других проблем у детей, которым поставлен диагноз «синдром Корнелии де Ланге». Симптомы заболевания, связанные с неврологическим состоянием ребенка, могут быть такими:
— новорожденные вяло сосут, часто срыгивают;
— малая подвижность и двигательная активность;
— гипотония мышц (мышечный тонус понижен, нет силы в руках и ногах);
— периодическое возникновение судорог.
Дети с синдромом де Ланге имеют проблемы со слухом, зрением и речью. Многие из них плохо говорят или почти не говорят в любом возрасте. Родители отмечают, что в большинстве случаев малыши свои желания выражают жестами. Со зрением у них бывают такие проблемы:
— косоглазие;
— близорукость;
— астигматизм;
— атрофия зрительного нерва.

Интеллектуальное развитие
Синдром Корнелии де Ланге кроме всех прочих осложнений со здоровьем вызывает умственную отсталость, которая отмечается практически у каждого больного ребенка, причем в 80 % случаев диагностируется имбецильность либо дебильность. Однако есть дети с синдромом де Ланге, которые посещают обычные дошкольные и школьные учреждения. Это зависит от того, в какой из двух форм диагностировано заболевание. Первая называется классической, при которой наблюдаются многие отклонения в формировании и работе внутренних органов, внешние аномалии и ярко выраженная задержка умственного развития. Вторая форма условно называется смазанной. При ней наблюдаются те или иные внешние отклонения, есть некоторые проблемы с внутренними органами, но интеллектуальное развитие имеет пограничную задержку.
Как отмечают родители, дети с синдромом де Ланге в любом возрасте не просятся в туалет, часто подвержены раздражению, любят выполнять различные несвойственные здоровым детям действия: постоянно рвать бумагу, есть ее, ломать все, что попадает им в руки, все время двигаться по кругу. Такие действия детей как бы их успокаивают.
Причины заболевания
Прошло практически сто лет с момента, когда впервые был описан синдром Корнелии де Ланге. Причины заболевания за это время удалось выяснить. Ими являются мутации в генах. Наибольшее число случаев зафиксировано при мутациях в 5-й хромосоме, точнее в гене NIPBL, находящемся в ее плече «р». Небольшой процент возникновения синдрома де Ланге зафиксирован у людей с мутацией 1А-белка хромосом (называется ген SMC1A), и один случай отмечен с мутацией также в белке хромосом, называемом геном SMC3. Однако причины, вызывающие эти генные мутации, пока находятся в стадии теорий и предположений.
Есть мнение, что их могут вызывать инфекционные заболевания беременной, особенно в первом триместре, некоторые лекарственные препараты, преклонный возраст отца ребенка либо возраст рожениц старше 35 лет, а также браки между родственниками.
Но ни одна из перечисленных причин не является бесспорной и 100 % вызывающей генетические изменения.
Еще прорабатывается гипотеза, что синдром Корнелии де Ланге передается по наследству, однако больше чем у половины обследованных пациентов это заболевание возникло спорадически, впервые в роду.
Синдром Корнелии де Ланге: прогноз
Считается, что данное заболевание очень редкое. Единых данных, с какой частотой оно встречается, нет. Одни источники утверждают, что заболевает один ребенок на 10 000 новорожденных, другие, что один на 100 000, третьи называют различные цифры в этом диапазоне. Если вдуматься, это не так уж и мало. Согласно статистике ежедневно на земле рождается около 370 тысяч младенцев. То есть, если взять даже самые низкие показатели, каждый день рождается примерно 4 человека, у которых диагностируется синдром Корнелии де Ланге.
Сколько живут такие люди, зависит от многих факторов, определяющими из которых являются степень аномалий внутренних органов, своевременное их выявление и качество оказанных медицинских воздействий. Если у ребенка патологии внутренних органов, несовместимые с жизнью, он умирает в первый месяц после рождения. Если же аномалии внутренних органов незначительны или ребенку было вовремя произведено хирургическое вмешательство, срок его жизни может быть достаточно долгим. Осложняет прогноз то обстоятельство, что организм больных с синдромом де Ланге не способен оказывать стойкое сопротивление обычным болезням, например вирусным, и труднее с ними борется.

Лечение
Большинство авторов и источников утверждают, что не существует возможности вылечить ребенка, у которого диагностирован синдром Корнелии де Ланге. Лечение сводится к хирургическим операциям (если есть показания), приему витаминов, ноотропных средств (воздействуют на функции мозга), анаболиков, проведении седативной терапии. Однако в наше время высочайших технологий можно если не полностью победить недуг, то значительно снизить его проявление. Для этого нужны вера в успех, нечеловеческое терпение и деньги, потому что лечение дорогое. Вот контакты клиник и центров, где берутся помочь:
1. Киев. Научно-методический центр «Истина», расположен на улице Вильямса, строение № 4. Телефоны: +38-044-467-63-89, +38-095-068-30-74. Здесь работают по методу Ульяны Лущик, есть много положительных отзывов.
2. Москва, Солнцево. Научно-практический центр, расположенный на улице Авиаторов, строение № 38. Телефоны: +7-495-934-17-53, +7-495-934-27-10, +7-495-934-14-39. Есть много отзывов, что детям с синдромом де Ланге после курсов лечения в этом центре становится гораздо лучше.
3. Израиль. Центр биокоррекции им. Васильева (лечение стоит от 10 тысяч у. е.). Телефон: 972-352-333-89.
У детей с диагнозом «синдром Корнелии де Ланге» продолжительность жизни во многом зависит от самоотверженной заботы о них родных людей, ведь заниматься с такими больными нужно практически каждую минуту. Очень часто достигнутые положительные результаты лечения уменьшаются или сводятся к нулю, если прекращают лечение или просто потому что происходит рецидив.
По отзывам родителей, положительные результаты в лечении их детей дают кинезотерапия, специальные реабилитационные программы плавания с дельфинами в дельфинарии, биоритмокоррекция, ароматерапия, музыкальные занятия, светотерапия.
Профилактика
Трудно говорить о профилактических мероприятиях заболевания, причины возникновения которого никому не известны. Учитывая установленные факторы, способные вызвать мутации на генетическом уровне, можно посоветовать:
— не допускать зачатия детей от кровных родственников;
— с осторожностью относиться к поздним материнству и отцовству;
— в период беременности, особенно в первые месяцы, принимать все меры, чтобы избежать вирусных инфекций, а в случае заболевания принимать лекарства только после консультации с врачом.
На основе проведенных исследований пациентов с синдромом Корнелии де Ланге врачи склонны считать, что в одной семье повторно может родиться больной ребенок в 2 % случаев, а в тех семьях, где у отца или у матери присутствуют некоторые симптомы синдрома де Ланге, больной ребенок может родиться в 25 % случаев. В связи с этим все женщины из группы риска должны проходить углубленную пренатальную диагностику, в частности проверять наличие в сыворотке крови белка РАРРА. Если он отсутствует, есть очень высокая вероятность родить ребенка с синдромом Корнелии де Ланге.
Источник
Синдром Корнелии де Ланге (синдром Брахмана-Ланге) — наследственное заболевание, проявляющееся умственной отсталостью и множественными аномалиями развития. Частота заболевания — примерно 1 на 10000[1].
Впервые случай такого заболевания описан в 1916 году немецким врачом В. Брахманом (W. R. C. Brachmann)[2], название синдром получил по имени голландского педиатра Корнелии де Ланге (Cornelia de Lange), в 1933 году описавшей синдром на основе анализа пяти случаев заболевания[3].
Генетика[править | править код]
Синдром Корнелии де Ланге относится к доминантно-наследуемым заболеваниям, большинство случаев являются спорадическими, возникшими de novo. Синдром является генетически гетерогенным. Примерно половина случаев обусловлена мутациями в гене NIPBL, около 5 % случаев — мутациями в гене SMC1A, кодирующем субъединицу белкового комплекса когезина. Один описанный случай связан с мутацией в гене SMC3, который также кодирует одну из субъединиц когезина[4].
В ряде случаев при цитогенетическом исследовании находят микродупликацию локусов q25 — q29 хромосомы 3[5].
Клиническая картина[править | править код]
- Микроцефалия (уменьшение размеров черепа более чем на 10 % возрастной нормы);
- Брахицефалия (укорочение черепа в сагиттальном направлении, в результате чего поперечный размер головы увеличивается, а продольный уменьшается);
- Тонкие сросшиеся брови; длинные загнутые ресницы; деформированные ушные раковины; маленький нос, открытые вперед ноздри, атрезия хоан; тонкая верхняя губа;
- Микрогения; высокое нёбо или расщелина нёба; нарушение прорезывания зубов;
- Миопия, косоглазие, астигматизм, атрофия зрительных нервов, колобома зрительного нерва;
- Маленькие кисти и стопы, отсутствие или значительное недоразвитие проксимальных отделов конечностей, вследствие чего кисти и стопы кажутся прикрепленными непосредственно к туловищу, уменьшение количества пальцев;
- Мраморная кожа;
- Гипоплазия сосков;
- Гипертрихоз;
- Судороги;
- Врожденные пороки внутренних органов (сердца, почек, пилоростеноз, крипторхизм) и др.[5]
У части больных наблюдается бег по кругу, стереотипные движения руками и самоповреждения[6].
У всех больных отмечаются отставание в росте, в 80 % случаев — тяжёлая умственная отсталость (имбецильность), в оставшихся 20 % менее выраженные интеллектуальные нарушения[6]; типичны рецидивирующие респираторные инфекции. Выделяют два варианта синдрома: первый (классический) с выраженной пренатальной гипоплазией, значительной задержкой физического и интеллектуального развития, грубыми пороками развития; второй — с аналогичными лицевыми и малыми скелетными аномалиями, но пограничной задержкой психомоторного развития и отсутствием грубых пороков развития[5].
Примечания[править | править код]
- ↑ Opitz J. M., Reynolds J. F. The Brachmann‐de Lange syndrome //American journal of medical genetics. — 1985. — Т. 22. — №. 1. — С. 89-102.
- ↑ Brachmann W. Ein Fall von symmetrischer Monodaktylie durch Ulnadefekt, mit symmetrischer Flughautbildung in den Ellenbeugen, sowie anderen Abnormitäten (Zwerghaftigkeit, Halsrippen, Behaarung): Aus d. Inn. Abt. d. Kinderheilanst. in Dresden; Leiter: Brückner : дис. — Krager, 1916.
- ↑ Cornelia de Lange. Sur un type nouveau de degeneration (typus Amstelodamensis). Archives de medecine des enfants, 1933; 36: 713—719
- ↑ Schrier S. A. et al. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature //American Journal of Medical Genetics Part A. — 2011. — Т. 155. — №. 12. — С. 3007-3024
- ↑ 1 2 3 Бородулин В.И., Тополянский А.В. и др. Синдромы и симптомы в клинической практике. Эпонимический словарь-справочник. — М: Эксмо, 2009. — 464 с. — ISBN 978-5-699-33854-2.
- ↑ 1 2 Исаев Д. Н. Умственная отсталость у детей и подростков. Руководство. — СПб: Речь, 2003. — С. 58. — 397 с. — ISBN 5-9268-0212-1.
Источник
В начале двадцатого века на заболевание обратил внимание немецкий врач В. Брахман. На примере наблюдаемого пациента он описал основные симптомы генетической патологии. Позже, в тридцатых годах того же столетия, голландский педиатр Корнелия де Ланге (де Лонга) в разное время вела пятерых пациентов, не состоящих в родстве и имеющих сходные признаки заболевания. По итогам исследования врач сделала заключение и опубликовала работу.
Синдром относится к редким генетически гетерогенным болезням, в равной степени поражает девочек и мальчиков, встречается на всех континентах. По статистическим данным, на 10–30 тысяч новорожденных приходится один случай с этой патологией. Еще одно название синдрома – Амстердамский нанизм (карликовость). В городе зафиксировано три индивида с врожденной аномалией. Болезнь протекает на фоне пороков развития скелета, внутренних органов, умственной отсталости.
Причины возникновения
Патогенезом аномалии является мутация генов NIPBL, SMC3, SMCIA, их белки участвуют в работе когезинового комплекса, присоединяя фермент к хромосоме в метафазе. Нарушение функционирования лежит в молекулярной основе синдрома. У половины пациентов определяется сломанный генетический код NIPBL, содержащий больше 40 экзонов, гетерозиготные мутации вызывают сбой в процессе de novo.

Были зафиксированы случаи передачи синдрома аутосомно-доминантным путем. Мозаичная форма NIPBL отмечалась у родителей в стадии эмбриона. Носители сломанного гена способны передать его потомкам через одно или через несколько поколений. У совершенно здоровой женщины и мужчины возможно рождение ребенка с синдромом Корнелии де Ланге.
В пяти процентах случаев аномалия формируется по вине мутации SMC1A, он также кодирует белки, участвующие в когезиновом процессе. Заболевание характеризуется легкой формой фенотипа. Нарушенный ген представляет опасность для всех поколений семьи, потому что напрямую связан с Х-хромосомой. Описан один пациент с изменением SMC3 с характерными чертами лица и строением черепа, при этом недоразвитость внутренних органов отсутствовала. В 40% случаев этиологию еще предстоит выяснить.
Обширный перечень симптомов
Определить наличие аномалии можно визуально сразу после рождения младенца. Ребенок появляется на свет с дефицитом веса (2/3 от общепринятой нормы). Синдром Корнелии де Ланге у пациента проявляется отличительными чертами в строении скелета, сопровождается неврологическим расстройством, отсталым умственным развитием.
Аномалии головы
Отклонение от критериев в строении лица и черепа является основным показателем для определения патологии.
Отмечается:
- объем меньше на 15%, чем у здорового новорожденного (микроцефалия);
- ширина больше продольного размера (брахицефалия);
- верхняя (мозговая) часть уменьшена.
Дефекты лица:
- четко очерченные брови, сросшиеся (сплошная линия), негустые, недоразвитые надбровные дуги;
- ресницы длинные, пушистые, загнутые кверху, разрез глаз – по монголоидному типу;
- переносица запавшая, широкая, нос маленький, с явно выраженными ноздрями;
- нижняя челюсть не пропорциональна лицу, маленькая, квадратной формы;
- губы тонкие, с опущенными уголками;
- редкие зубы (олигодонтия) или полное их отсутствие;
- небо высокое, без перегородки (волчья пасть).
В 50% случаев выявляется повышенная волосистость тела в основном в области спины, низкая линия роста волос на лбу и затылке. Кожный покров имеет синюшный оттенок, хорошо просматривается сетка сосудов (мраморность). Последний признак не относится к основным показателям синдрома.
Недоразвитость внутренних органов
Аномалии внутренних органов являются серьезной угрозой для жизни индивида. При заболевании отмечается:
- закрытость носовых проходов (атрезия хоан), пациент дышит при помощи зонда;
- пороки со стороны сердца: дефекты в структуре сосудистой системы, перегородок, клапана, отверстие в предсердии;
- нарушения желудочно-кишечного тракта: сужение привратника, подвижность слепой кишки, грыжа на диафрагме, желудочный рефлюкс, стеноз пищевода;
- аномалии развития в мочевыводящей системе;
- недоразвитость половых органов: маленький половой член, пустая мошонка (отсутствие яичек), крипторхизм, двурогая матка;
- новообразование на почках, гидронефроз.
Также патологию сопровождают дефекты в мозговой ткани, характеризующиеся аплазией мозолистого тела и дисплазией извилин.
Костно-мышечный аппарат
При синдроме отмечается патологическое строение конечностей:
- укороченные руки по отношению к телу, отсутствие предплечий;
- неподвижность локтевого сустава (анкилоз) в результате образования хряща;
- сращивание нескольких пальцев (синдактилия), искривление пятого (клинодактилия);
- фаланги квадратной формы, приплюснуты;
- фокомелия, развитые ступни и кисти на фоне микромелии (короткие руки и ноги);
- отсутствие бугорков мышц на мизинце и большом пальце;
- образование глубоких складок на ладони;
- нижние конечности подвержены аналогичным изменениям.
Дефекты в развитии позвоночника, рост ниже нормы, соответствующей году ребенка, деформированная грудная клетка, короткая шея.
Неврологические отклонения
Проявление болезни характеризуется следующими нарушениями нервной системы:
- косоглазие, близорукость, атрофия глазного нерва, птоз (опущение верхнего века);
- гипертонус мышц, судороги;
- нарушение в работе сегментарного аппарата, которое приводит к повышению рефлексов;
- расположение рук на уровне глаз;
- хождение по кругу;
- нарушение сна;
- частичная потеря слуха или глухота;
- склонность к самоповреждению, депрессии.
У ребенка с возрастом развивается синдром дефицита внимания, он не может длительное время заниматься одним делом, концентрация отсутствует, проявляются признаки гиперактивности, отмечается невроз навязчивых состояний.
Умственное развитие
Неврологические отклонения и аномальные изменения в головном мозге влияют на умственное развитие, способствуют проявлению имбецильности. В легкой форме патологии пациент способен к обучению, словарный запас находится в рамках бытового общения. Такие индивиды посещают детские учреждения, но при этом не обладают критическим мышлением, трудно сходятся со сверстниками, возможно проявление аутизма.
Более тяжелая форма сказывается на коммуникативных способностях. Адаптация к социуму проходит в специализированных учреждениях. Склонность к проявлению агрессии. Невозможность самостоятельного существования в незнакомой обстановке. Пациенты неспособны к самообслуживанию, требуют ухода со стороны близких людей. Тяжело поддаются обучению. С годами процесс деградации только усугубляется.
Диагностика
Определить наличие болезни у эмбриона при помощи современной диагностики практически невозможно. Показателем развития синдрома является низкая концентрация в плазме крови РАРР-А, этот вид протеина у женщины в период вынашивания ребенка производится в большом количестве. Обнаружить хромосомное отклонение у плода можно только с условием уменьшения показателя РАРР-А. Поэтому определяется патология после рождения младенца по визуальным признакам.

При наличии хотя бы одного симптома назначается инструментальная диагностика для выявления дефектов развития внутренних органов, несовместимых с жизнью. Методика включает:
- ультразвуковое обследование;
- магниторезонансную томографию;
- риноскопию;
- рентгенографию.
Назначаются клинические и цитогенетические анализы.
Необходим дифференциальный подход, чтобы исключить синдром, похожий на Корнелия де Ланге. Тяжелый фенотип имеет сходство с проявлением аутосомно-рецессивным заболеванием Фринса, которому свойственны такие признаки: грыжа диафрагмы, изменение лица, волчья пасть, недоразвитые конечности. Различием является заячья губа и макростомия (большой вес плода). Новорожденный с алкогольным синдромом также внешне похож на больного с Амстердамским нанизмом. Это объясняется задержкой внутриутробного развития. Различием является нормальный размер конечностей и расположение фаланг. В этом случае установить диагноз поможет семейный анамнез и хромосомный анализ.
Продолжительность жизни
При синдроме Корнелии де Ланге продолжительность жизни зависит от степени дефекта развития внутренних органов. Если аномалия несовместима с жизнью и своевременно не проведено оперативное вмешательство, смерть может наступить в течение месяца после рождения. Отсутствие соматических отклонений, ранняя диагностика дефектов, позволяющая их устранить, обеспечивает долгую жизнь пациентам. Прогноз ухудшается в случае вирусных инфекций, которым неспособна противостоять иммунная система. Обычный грипп может закончиться летальным исходом. В среднем дети с синдромом Корнелии де Ланге доживают до 13 лет. Фиксировались случаи при легкой, стертой форме, после адекватного устранения аномалий, когда пациент умирал в преклонном возрасте.
Лечение и профилактика синдрома Корнелии де Ланге
Специфического воздействия на патологию не существует. Терапия носит симптоматичный характер, основной упор делается на хирургическое вмешательство. После оценки питания и роста, если ребенок не может есть из бутылочки, рекомендуется подача калорий с помощью назогастрального зонда. При желудочном рефлюксе показано создание гастростомы. Применение гормона роста нецелесообразно из-за резистентности к препарату.
Медикаментозное лечение эффективно при дефиците внимания, назначается «Метилфенидат» с его помощью нормализуется гиперактивность. Для купирования тревожного состояния применяется «Буспирон». Стабилизирует настроение «Сертралин» – ингибитор обратного захвата гормона серотонина. Мышечные спазмы снимаются противосудорожными препаратами oкcaзoлидиoнoнoвой группы.
При офтальмологических дефектах проводится хирургическая коррекция птоза, лечение конъюнктивы, ношение очков. Рекомендуется своевременное купирование отитов стандартным набором антибиотиков, устранение заболеваний среднего уха, установка слухового аппарата. Пороки сердца удаляются оперативным путем. Аномалии, связанные с почками и мочевыделительной системой, убираются традиционными консервативными методами.
Профилактические меры
С учетом неточной картины патогенеза рекомендации, предупреждающие заболевание, призваны не допустить наследственной передачи мутированного гена. Профилактические мероприятия заключаются:
- в обязательном обследовании беременной женщины на уровень протеина-А в крови;
- не допускать инфекционных заболеваний особенно в период первого триместра;
- предотвращение зачатия путем инцеста (родители ребенка – кровные родственники);
- постоянное наблюдение у врача беременной женщины после сорока лет, обследование мужчины в случае позднего отцовства.
Рекомендуется медико-генетическая консультация перед зачатием родителю, у которого в семейном анамнезе есть прецеденты синдрома Корнелии де Ланге.
Загрузка…
Источник