Синдром клиппеля треноне вебера википедия

Klippel–Trénaunay syndrome formerly Klippel–Trénaunay–Weber syndrome[1] and sometimes angioosteohypertrophy syndrome and hemangiectatic hypertrophy,[2] is a rare congenital medical condition in which blood vessels and/or lymph vessels fail to form properly. The three main features are nevus flammeus (port-wine stain), venous and lymphatic malformations, and soft-tissue hypertrophy of the affected limb.[2] It is similar to, though distinctly separate from, the less common Parkes-Weber syndrome.
The classical triad of Klippel-Trenaunay syndrome consists of:[3]
- vascular malformations of the capillary, venous and lymphatic vessels;
- varicosities of unusual distribution, particularly the lateral venous anomaly; and
- unilateral soft and skeletal tissue hypertrophy, usually the lower extremity.
Signs and symptoms[edit]
The birth defect is diagnosed by the presence of a combination of these symptoms (often on approximately 1⁄4 of the body, though some cases may present more or less affected tissue):
- One or more distinctive port-wine stains with sharp borders
- Varicose veins
- Hypertrophy of bony and soft tissues, that may lead to local gigantism or shrinking, most typically in the lower body/legs.
- An improperly developed lymph system
In some cases, port-wine stains (capillary port wine type) may be absent. Such cases are very rare and may be classified as «atypical Klippel–Trenaunay syndrome».
KTS can either affect blood vessels, lymph vessels, or both. The condition most commonly presents with a mixture of the two. Those with venous involvement experience increased pain and complications, such as venous ulceration in the lower extremities.
Those with large AVMs are at risk of formation of blood clots in the vascular lesion, which may migrate to the lungs (pulmonary embolism). If there is large-volume blood flow through the lesion, high-output heart failure may develop due to the inability of the heart to generate sufficient cardiac output.[4] Much rarer are haemorrhages. They can be serious if the brain is affected.[5]
| Symptom | % |
|---|---|
| Port wine stain | 98% |
| Longer extremity/hypertrophy | 67% |
| Circumferential hypertrophy | 77% |
| Varicosity | 72% |
| Pain | 37% |
| Bleeding | 17% |
| Superficial thrombophlebitis | 15% |
| Cellulitis | 13% |
| Rectal bleeding | 12% |
| Lymphatic malformation | 11% |
| Lymphedema | 10% |
| Hyperpigmentation | 8% |
| Ankle ulcers | 6% |
| Verrucae | 6% |
| Hyperhidrosis | 5% |
| DVT | 4% |
| Induration | 4% |
| Pulmonary embolism | 4% |
| Limb numbness | 2% |
Genetics[edit]
The birth defect affects men and women equally, and is not limited to any racial group. It is not certain if it is genetic in nature, although testing is ongoing.[7] There is some evidence that it may be associated with a translocation at t(8;14)(q22.3;q13).[8] Some researchers have suggested AGGF1 has an association.[9]
Diagnosis[edit]
Differential diagnosis[edit]
- Parkes Weber syndrome
- Proteus syndrome[10]
- Macrodystrophia lipomatosa[11]
Classification[edit]
There is disagreement as to how cases of KTS should be classified if there is an arteriovenous fistula present. Although several authorities have suggested that the term Parkes-Weber syndrome is applied in those cases,[4][12][13]ICD-10 currently uses the term «Klippel–Trénaunay–Weber syndrome».
Treatment[edit]
KTS is a complex syndrome, and no single treatment is applicable for everyone. Treatment is decided on a case-by-case basis with the individual’s doctors.
At present, many of the symptoms may be treated, but there is no cure for Klippel–Trenaunay syndrome.[14]
Surgical[edit]
Debulking has been the most common treatment for KTS for several decades and while improvements have been made, the procedure is still considered invasive and has several risks associated with it. More effective and less invasive treatment choices now exist for KTS patients and therefore debulking is generally only recommended as a last resort. Debulking operations can result in major deformities and also leave patients with permanent nerve damage.[citation needed]
Mayo Clinic has reported the largest experience in managing KTS with major surgery. In 39 years at Mayo clinic the surgery team evaluated 252 consecutive cases of KTS, of which only 145 (57.5%) could be treated by primary surgery.[15] The immediate success rate for treating varicose veins was only 40%, excision of vascular malformation was possible in 60%, debulking operations in 65%, and correction of bone deformity and limb length correction (epiphysiodesis) had 90% success. All the procedures demonstrated high recurrence rate in the follow-up. Mayo clinic studies demonstrate that primary surgical management of KTS has limitations and non-surgical approaches need to be developed in order to offer a better quality of life for these patients. Major surgery including amputation and debulking surgery does not seem to offer any benefit on a long-term basis.[citation needed]
Nonsurgical[edit]
Sclerotherapy is a treatment for specific veins and vascular malformations in the affected area. It involves the injection of a chemical into the abnormal veins to cause thickening and obstruction of the targeted vessels. Such treatment may allow normal blood flow to resume. It is a non-surgical medical procedure and is not nearly as invasive as debulking. Ultrasound guided foam sclerotherapy is the state of the art new treatment which could potentially close many large vascular malformations.[16][17]
Compression therapies are finding more use as of the last ten years. The greatest issue with KTS syndrome is that the blood flow and/or lymph flow may be impeded, and will pool in the affected area. This can cause pain, swelling, inflammations, and in some cases, even ulceration and infection. Among older children and adults, compression garments can be used to alleviate almost all of these, and when combined with elevation of the affected area and proper management, can result in a comfortable lifestyle for the patient without any surgery. Compression garments are also used lately after a debulking procedure to maintain the results of the procedure. For early treatment of infants and toddlers with KTS, custom compression garments are impractical because of the rate of growth. When children may benefit from compression therapies, wraps and lymphatic massage may be used. While compression garments or therapy are not appropriate for everyone, they are relatively cheap (compared to surgery), and have few side-effects. Possible side-effects include a slight risk that the fluids may simply be displaced to an undesirable location (e.g., the groin), or that the compression therapy itself further impedes circulation to the affected extremities.[citation needed]
History[edit]
The condition was first described by French physicians Maurice Klippel and Paul Trénaunay in 1900; they referred to it as naevus vasculosus osteohypertrophicus.[18][19] The German-British physician Frederick Parkes Weber described cases in 1907 and 1918 that were similar but not identical to those described by Klippel and Trénaunay.[20][21]
References[edit]
- ^ «Klippel-Trenaunay syndrome». Archived from the original on July 4, 2013. Retrieved May 15, 2014.
- ^ a b James, William; Berger, Timothy; Elston, Dirk (2005). Andrews’ Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. p. 585. ISBN 978-0-7216-2921-6.
- ^ Karim, Tanweer; Nanda, NavdeepS; Singh, Upvan (2014). «A rare presentation of Klippel-Trenaunay syndrome». Indian Dermatology Online Journal. 5 (2): 154–6. doi:10.4103/2229-5178.131086. PMC 4030342. PMID 24860749.
- ^ a b Mendiratta, V; Koranne, RV; Sardana, K; Hemal, U; Solanki, RS (2004). «Klippel trenaunay Parkes-Weber syndrome». Indian Journal of Dermatology, Venereology and Leprology. 70 (2): 119–22. PMID 17642585.
- ^ Petzold, A.; Bischoff, C.; Conrad, B. (2000). «Repetitive cerebral bleeding in an adult with Klippel-Trenaunay syndrome». Journal of Neurology. 247 (5): 389–391. doi:10.1007/s004150050609. PMID 10896274.
- ^ Klippel-Trenaunay syndrome: Spectrum and management
- ^ Tian XL, Kadaba R, You SA, Liu M, Timur AA, Yang L, Chen Q, Szafranski P, Rao S, Wu L, Housman DE, DiCorleto PE, Driscoll DJ, Borrow J, Wang Q (2004). «Identification of an angiogenic factor that when mutated causes susceptibility to Klippel–Trenaunay syndrome» (PDF). Nature. 427 (6975): 640–5. Bibcode:2004Natur.427..640T. doi:10.1038/nature02320. PMC 1618873. PMID 14961121. Archived from the original (PDF) on December 9, 2006.
- ^ Wang, Q.; Timur, A.A.; Szafranski, P.; Sadgephour, A.; Jurecic, V.; Cowell, J.; Baldini, A.; Driscoll, D.J. (2001). «Identification and molecular characterization of de novo translocation t(8;14)(q22.3;q13) associated with a vascular and tissue overgrowth syndrome». Cytogenetic and Genome Research. 95 (3–4): 183–8. doi:10.1159/000059343. PMC 1579861. PMID 12063397.
- ^ Barker, K T; Foulkes, WD; Schwartz, CE; Labadie, C; Monsell, F; Houlston, RS; Harper, J (2005). «Is the E133K allele of VG5Q associated with Klippel-Trenaunay and other overgrowth syndromes?». Journal of Medical Genetics. 43 (7): 613–4. doi:10.1136/jmg.2006.040790. PMC 2564558. PMID 16443853.
- ^ EL-Sobky TA, Elsayed SM, EL Mikkawy DME (2015). «Orthopaedic manifestations of Proteus syndrome in a child with literature update». Bone Rep. 3: 104–108. doi:10.1016/j.bonr.2015.09.004. PMC 5365241. PMID 28377973.CS1 maint: multiple names: authors list (link)
- ^ Abdulhady, H; El-Sobky, TA; Elsayed, NS; Sakr, HM (11 June 2018). «Clinical and imaging features of pedal macrodystrophia lipomatosa in two children with differential diagnosis review». Journal of Musculoskeletal Surgery and Research. 2 (3): 130. doi:10.4103/jmsr.jmsr_8_18.
- ^ Lindenauer, S. Martin (1965). «The Klippel-Trenaunay Syndrome». Annals of Surgery. 162 (2): 303–14. doi:10.1097/00000658-196508000-00023. PMC 1476812. PMID 14327016.
- ^ Cohen, M. Michael (2000). «Klippel-Trenaunay syndrome». American Journal of Medical Genetics. 93 (3): 171–5. doi:10.1002/1096-8628(20000731)93:3<171::AID-AJMG1>3.0.CO;2-K. PMID 10925375.
- ^ Black, Rosemary (May 19, 2009). «What is Klippel–Trenaunay Syndrome? Brooklyn writer Carla Sosenko shares facts about condition». New York Daily News.
- ^ Jacob, A G; Driscoll, D J; Shaughnessy, W J; Stanson, A W; Clay, R P; Gloviczki, P (1998). «Klippel-Trenaunay syndrome: Spectrum and management». Mayo Clinic Proceedings. 73 (1): 28–36. doi:10.4065/73.1.28. PMID 9443675.
- ^ Cabrera, Juan; Cabrera Jr, J; Garcia-Olmedo, MA; Redondo, P (2003). «Treatment of Venous Malformations with Sclerosant in Microfoam Form». Archives of Dermatology. 139 (11): 1409–16. doi:10.1001/archderm.139.11.1409. PMID 14623700.
- ^ McDonagh, B; Sorenson, S; Cohen, A; Eaton, T; Huntley, D E; La Baer, S; Campbell, K; Guptan, R C (2005). «Management of venous malformations in Klippel–Trenaunay syndrome with ultrasound-guided foam sclerotherapy». Phlebology. 20 (2): 63–81. doi:10.1258/0268355054069188.
- ^ synd/1812 at Who Named It?
- ^ Klippel M, Trénaunay P (1900). «Du naevus variqueux ostéohypertrophique». Archives Générales de Médecine. 3: 641–72.
- ^ Weber FP (1907). «Angioma-formation in connection with hypertrophy of limbs and hemi-hypertrophy». British Journal of Dermatology. 19: 231–5.
- ^ Weber FP (1918). «Hemangiectatic hypertrophy of limbs – congenital phlebarteriectasis and so-called congenital varicose veins». British Journal of Children’s Diseases. 25: 13.
External links[edit]
Источник
- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Игушева Н.А.
1
Кузнецова В.В.
1
1 ФГБОУ ВО ПГМУ им. академика Е.А. Вагнера Минздрава России
Синдром Клиппеля ‒ Треноне ‒ Вебера ‒ состояние, влияющее на развитие кровеносных сосудов, кожи, мышц и костей. Данное расстройство имеет характерные черты в виде классической триады: капиллярную гемангиому, называемую винным пятном, гипертрофию мягких тканей и костей, пороки развития вен. Это состояние было впервые описано французскими врачами Морисом Клиппелем и Паулем Треноне в 1900 году, которое они его называли «nevus vasculosus osteohypertrophicus». Немецко-британский врач Фредерик Паркес Вебер описал случаи 1907 и 1918 годов, которые были похожи, но не идентичны тем, которые описывали Клиппель и Треноне. Врожденный дефект поражает мужчин и женщин в равной степени и не ограничивается в пределах какой-либо расовой группы. Не имеется абсолютного подтверждения, что патология носит генетический характер, хотя изучение ее все еще продолжается.
синдром Клиппеля ‒ Треноне ‒ Вебера
винное пятно
гипертрофия мягких тканей и костей
пороки развития вен
генная мутация
лимфостаз
PIK3CA
р110α
PI3K.
1. Klippel-Trenaunay-Weber Syndrome: Genetics home reference. ‒ 1995 [Электронный ресурс]. URL: https://ghr.nlm.nih.gov/condition/klippel-trenaunay-syndrome#sourcesforpage (дата обращения: 24.02.2018)
2. Klippel-Trenaunay-Weber Syndrome: Cardiology news & opinion. ‒ 1994 [Электронный ресурс]. URL: https://reference.medscape.com/article/1084257-differential (дата обращения 25.02.2018).
3. Luks VL, Kamitaki N, Vivero MP, Uller W, Rab R, Bovée JV, Rialon KL, Guevara CJ, Alomari AI, Greene AK, Fishman SJ, Kozakewich HP, Maclellan RA, Mulliken JB, Rahbar R, Spencer SA, Trenor CC 3rd, Upton J, Zurakowski D, Perkins JA, Kirsh A, Bennett JT, Dobyns WB, Kurek KC, Warman ML, McCarroll SA, Murillo R. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015 Apr;166(4):1048-54.
4. Sfaihi L, Aissa K, Fourati H, Kamoun F, Mnif Z, Kamoun T, et al. Klippel Trenaunay syndrome in association with Sturge Weber syndrome about one case. Tunis Med. 2016 Feb. 92(2):173-4.
5. Sung HM, Chung HY, Lee SJ, Lee JM, Huh S, Lee JW, et al. Clinical Experience of the Klippel-Trenaunay Syndrome. Arch Plast Surg. 2015 Sep. 42 (5):552-8.
6. Upadhyay H, Sherani K, Vakil A, Babury M. A case of recurrent massive pulmonary embolism in Klippel-Trenaunay-Weber syndrome treated with thrombolytics. Respir Med Case Rep. 2016. 17:68-70.
7. Vahidnezhad H, Youssefian L, Uitto J. Klippel-Trenaunay syndrome belongs to the PIK3CA-related overgrowth spectrum (PROS). Exp Dermatol. 2016 Jan;25(1):17-9.
История
В 1900 году известные французские медики Клиппель и Треноне впервые описали синдром у 2-х пациентов с винным пятном, варикозным расширением вен конечности, а также с гипертрофией костных и мягких тканей пораженных конечностей. Они назвали синдром «naevus vasculosus osteohypertrophicus». В 1907 году Паркс Вебер, не подозревая об открытии Клиппеля и Треноне, описал пациента с тремя вышеупомянутыми симптомами, а также с артериовенозной мальформацией пораженной конечности. Он назвал этот процесс гемангиэктатической гипертрофией [1].
Определение и клиника
Синдром Клиппеля — Треноне — Вебера — состояние, влияющее на развитие кровеносных сосудов, кожи, мышц и костей. Данное расстройство имеет характерные черты в виде классической триады: капиллярную гемангиому, называемую винным пятном, гипертрофию мягких тканей и костей, пороки развития вен [4].
Большинство людей с синдромом Клиппеля — Треноне — Вебера с рождения имеют винное пятно, которое появляется в результате отека мелких кровеносных сосудов вблизи поверхности кожи [2]. Винные пятна плоские, имеют четкие границы, варьируются от бледно-розового до темно-бордового цвета, обычно локализуются на латеральной части одной конечности. С возрастом пораженный участок может стать светлее или темнее. Глубина гемангиомы может быть различной: она может быть ограничена кожей или проникать в низ лежащие слои, включающие мышцы и кости [1]. Висцеральные органы, такие как селезенка, печень, мочевой пузырь и толстая кишка, а также плевра, тоже могут быть вовлечены в патологический процесс [7].
Гипертрофия костных и мягких тканей является третьим признаком синдром Клиппеля — Треноне — Вебера. Гипертрофия конечности может быть вторичной по отношению к увеличенной длине (костное участие) и / или увеличению обхвата (вовлечение мягких тканей) [3]. Гипертрофия может быть оценена при рождении, а также в первые годы жизни. Большую степень гипертрофии можно наблюдать у пациентов с имеющейся у них артериовенозной мальформацией [1].
Обычно паталогический рост ограничен одной конечностью, чаще всего ногой, однако, разрастание также может затронуть руки, реже шею, голову и поясницу [5]. Аномальный рост может вызывать боль, чувство тяжести, снижение объема активных движений в зоне поражения. Также чрезмерный рост одной ноги приводит к проблемам с ходьбой. Иногда вовлеченная в процесс конечность может быть атрофирована, а не гипертрофирована [6].
Пороки развития вен являются третьей важной особенностью клинического течения синдрома Клиппеля — Треноне — Вебера. В эту группу аномалий входят варикозные расширения вен (обычно на латеральных сторонах бедер и на икрах ног), которые иногда замечаются при рождении [7]. Варикоз может не визуализироваться до тех пор, пока ребенок не начнет ходить. Варикозы могут быть обширными, хотя чаще всего они локализуются в пределах подкожной вены ноги [2].
Глубокие вены конечностей также могут вовлекаться в патологический процесс, повышая риск возникновения тромбоза, называемого тромбозом глубоких вен. Впоследствии оторвавшийся тромб, проходя с током крови через легкие, может привести к развитию легочной эмболии. В редких случаях варикозное расширение сосудов наблюдаются в мочевом пузыре, толстой кишке и легких [4].
Варикозы могут остаться стабильными в размере или постепенно расширяться, о чем будут свидетельствовать появление болей и лимфостаз. Эти симптомы могут ухудшиться во время беременности [1].
Другими особенностями клинического течения этого синдрома являются: лимфатическая обструкция, расщепление позвоночника, гипоспадия, синдактилия, олигодактилия, полидактилия, гипергидроз, гипертрихоз, парестезии, декальцификация костей, хроническая венозная недостаточность, застойный дерматит, плохое заживление ран, изъязвление, тромбоз, ангиосаркома. Орофасциальные аномалии могут потребовать специализированного ухода за зубами и анестезию [2].
Эпидемиология
Синдромом Клиппеля — Треноне — Вебера страдает не менее 1 на 100 000 человек по всему миру. Нет подтверждения о том, что данная патология развивается у представителей определенной расы. Страдают как мужчины, так и женщины в равной степени [3].
Патофизиология
Точная причина синдрома Клиппеля — Треноне — Вебера неизветна, хотя существуют несколько теорий. Близнак и Стэпл предложили теорию внутриутробного повреждения симпатических ганглиев или латерального промежуточного тракта, что приводит к дилатации микроскопических артериовенозных анастомозов [3]. Сервелль считал, что аномалии развития глубоких вен, с результирующей обструкцией венозного потока, приводят к венозной гипертензии, развитию варикоза и гипертрофии конечностей [6]. МакГрори и Амадио полагали, что лежащая в основе смешанная мезодермальная и эктодермальная дисплазия, вероятно, ответственны за развитие данного синдрома [7].
Синдром Клиппеля — Треноне — Вебера почти всегда является спорадическим, что означает, что он возникает у людей, не имеющих данной патологии в своей семье [2]. Исследования показывают, что это состояние является следствием генных мутаций, которые не наследуются. Эти генетические изменения, вызываемые соматическими мутациями, возникают случайным образом в одной клетке на ранних стадиях развития до рождения. По мере того как клетки продолжают делиться в период внутриутробного развития организма, одни дочерние клетки, возникшие от мутировавшей материнской, будут иметь мутации, а другие нет. Эта смесь клеток с генетической мутацией и без нее именуется понятием мозаицизм [1].
Также Кихичаком было высказано предположение, что синдром Клиппеля — Треноне — Вебера может быть вызван мутацией гена PIK3CA. Этот ген кодирует белок р110α, который является субъединицей фермента, называемого фосфатидилинозитол-3-киназа (PI3K). PI3K играет важную роль в клеточной жизнедеятельности, в том числе влияние на их пролиферацию, миграцию и выживаемость.
При возникновении мутаций в гене PIK3CA, подвергается изменению как белок p110α, так и фермент PI3K, вследствие чего у последнего неестественным образом повышается активность, позволяющая клеткам непрерывно расти и делиться. Повышенная клеточная пролиферация приводит к патологическому росту мягких тканей, костей и кровеносных сосудов [1].
Осложнения
- Осложнениями гемангиом являются повреждение и изъязвление кожи, кровотечение и присоединение вторичной инфекции [4].
- Варикозное расширение вен приводит к развитию парестезий, венозных язв, тромбоэмболии легочной артерии, тромбофлебита, застойного дерматита, кровоизлияний, гнойного воспаления подкожной клетчатки, отеков, вызванных лимфостазом [7].
- Гипертрофия конечности может привести к последующему сколиозу позвонков, нарушениям походки [3].
- Реже, осложнениями данной патологии являются срастания некоторых пальцев рук или ног (синдактилия) или увеличение их числа (полидактилия) [6].
- Также у пациентов с этим синдромом имеется склонность к развитию дегенеративных заболеваний суставов в раннем возрасте [1].
Библиографическая ссылка
Игушева Н.А., Кузнецова В.В. СИНДРОМ КЛИППЕЛЯ ‒ ТРЕНОНЕ ‒ ВЕБЕРА // Международный студенческий научный вестник. – 2018. – № 4-2.;
URL: https://eduherald.ru/ru/article/view?id=18506 (дата обращения: 12.05.2020).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)
Источник
Как известно, врожденные патологии передаются на генетическом уровне от родителей или возникают из-за нарушения эмбриогенеза. Аномалии могут формироваться из любых тканей или органов. Чаще всего они обнаруживаются сразу после рождения ребенка, но иногда проявляются гораздо позже. Примером врожденной аномалии является синдром Клиппеля-Вебера-Треноне. Данное заболевание относится к патологиям сосудистой системы, поражает преимущественно вены нижних конечностей.
В отличие от многих других врожденных аномалий, синдром Клиппеля-Вебера-Треноне может иметь благоприятный прогноз для жизни. Он достигается с помощью своевременного хирургического лечения недуга.
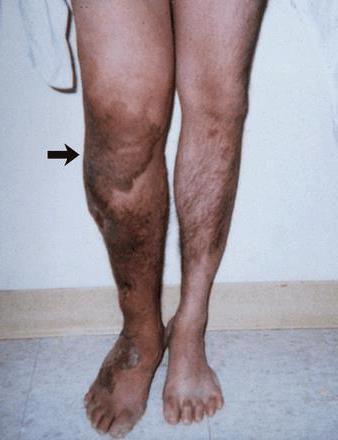
Патология: синдром Клиппеля-Треноне-Вебера – что это?
Данное заболевание известно с начала 20 века, когда впервые было описано двумя учеными – Треноне и Клиппелем. Позже доктор Вебер дополнил уже имевшиеся сведения об этой патологии. Известно, что недуг имеет и другое название – остеогипертрофический венозный невус.
Синдром Клиппеля-Вебера-Треноне встречается в основном среди мужского населения. На данный момент сведений об основной причине развития заболевания и его эпидемиологии недостаточно. Основными симптомами патологии являются обширные «родимые пятна» (невусы) на ногах, различный диаметр нижних конечностей, варикоз. Несмотря на раннее развитие клинических признаков и тяжесть поражения, в некоторых случаях синдром поддается лечению. Это преимущество достигнуто благодаря появлению новых технологий в области сосудистой хирургии.
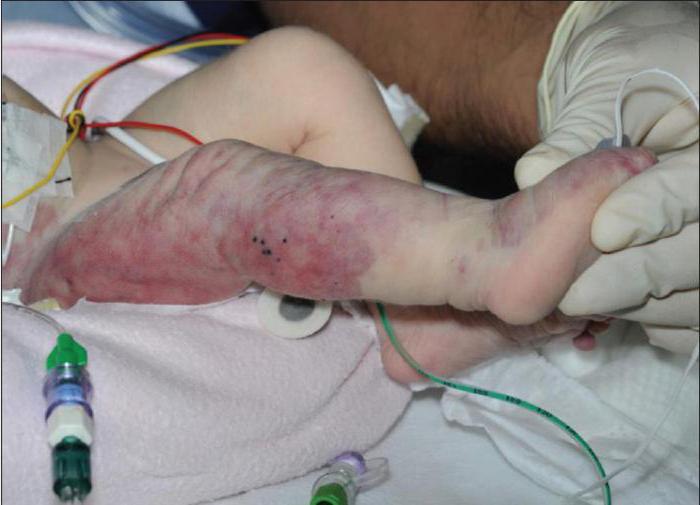
Причины проблемы
Так как заболевание было описано сравнительно недавно (около 100 лет назад), на данный момент нет достоверных сведений о его этиологии. Тем не менее существует несколько теорий, согласно которым развивается синдром Клиппеля-Треноне-Вебера. Причины патологии связывают с наследственной отягощенностью. Имеется в виду, что основным этиологическим фактором является мутация генов. Согласно другой теории, причина заболевания кроется в нарушении эмбриогенеза на ранних сроках беременности. Под действием неблагоприятных факторов окружающей среды (химические отравления, психоэмоциональные нагрузки, облучение радиацией) происходит аплазия венозной системы. Ее недоразвитие также связывают с инфекционными агентами. Согласно этой теории, причиной патологии являются бактерии или вирусы, попавшие в организм матери во время беременности.
Клиническая картина заболевания
Первые проявления синдрома Клиппеля-Треноне-Вебера заметны уже в период новорожденности. В редких случаях они возникают позже – в детском возрасте. Классическими симптомами заболевания являются следующие признаки:
- Ангиомы – сосудистые пятна. Чаще всего они имеются на одной ноге и охватывают большую площадь. Цвет ангиом может быть различным: от светлого до темного фиолетового оттенка. Под воздействием физических факторов (трение, удар) кожа в области «родимых пятен» легко повреждается, появляется кровоточивость.
- Варикозное расширение поверхностных вен ноги. Клинические проявления этого признака – извитость, утолщение, увеличение и болезненность сосудов нижней конечности.
- Гипертрофия пораженной ноги. Из-за поражения глубоких вен и их разрастания конечность увеличивается в диаметре. В некоторых случаях в процесс включена и костная система. Тогда пораженная нога может быть длиннее, чем здоровая конечность.
Иногда один из признаков заболевания отсутствует или не проявляется внешне (например, варикозное расширение поверхностных вен). В некоторых случаях патология быстро прогрессирует, и поражение переходит с конечностей на туловище и верхний плечевой пояс.
Диагностика
Заподозрить синдром Клиппеля-Вебера-Треноне можно по обширным ангиомам. Это симптом появляется в первую очередь. Позже присоединяются варикозная болезнь и гипертрофия конечности. Характерный признак данной патологии – одностороннее поражение. При подозрении на заболевание проводят лабораторную и инструментальную диагностику. В первую очередь выполняются УЗИ и допплерография нижних конечностей. Особенностью данного синдрома является отсутствие корреляции между венозным давлением и сердечной деятельностью. Этот признак отражается прямой линией при проведении венографии.
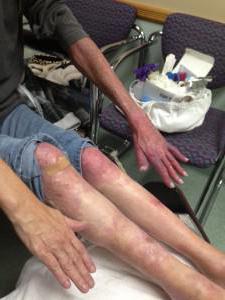
Синдром Клиппеля-Треноне-Вебера: лечение патологии
Устранение заболевания зависит от степени поражения глубоких вен. При значительном сужении проводится шунтирование. Если магистральная вена поражена на большом участке, то осуществляется трансплантация сосуда искусственным материалом. В качестве дополнительного лечения применяется склеротерапия (препараты «Тромбовар», «Фибро-Вейн»), ношение компрессионного белья. Также необходим постоянный прием антикоагулянтов (медикамент «Варфарин»). В некоторых случаях операцию на сосудах можно выполнить при помощи лазера.
Источник