Синдром каудальной регрессии мкб 10

Рубрика МКБ-10: Q76.0
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q65-Q79 Врожденные аномалии пороки развития и деформации костно-мышечной системы / Q76 Врожденные аномалии (пороки развития) позвоночника и костей грудной клетки
Определение и общие сведения[править]
Spina bifida occulta — скрытая щель дуг позвонков, чаще в области I крестцового позвонка.
Этиология и патогенез[править]
Клинические проявления[править]
Наиболее частая локализация этой формы — крестцовый и поясничный отделы позвоночника. На уровне незаращения дужек позвонков можно наблюдать различные патологические образования в виде плотных фиброзных тяжей, хрящевой и жировой ткани, липом, фибром и др.
Зачастую эта форма патологии настолько умеренно выражена, что не вызывает каких-либо беспокойств.
Spina bifida occulta: Диагностика[править]
При рентгенографии выявляют незаращение дужек, иногда и тел позвонков.
Дифференциальный диагноз[править]
Spina bifida occulta: Лечение[править]
Это состояние может потребовать хирургическое вмешательство.
Сейчас может применяться пренатальная хирургическая коррекция, как одна из областей применения фетальной хирургии.
Проведение внутриутробной операции по пластике спинномозговой грыжи снижает тяжесть и частоту осложнений, уменьшает вероятность постановки вентрикуло-перитонеального шунта, и значительно увеличивает шансы на полноценное физическое развитие ребенка.
Профилактика[править]
Прочее[править]
Синдром каудальной регрессии
Синонимы: каудальная дисплазия, синдром сакрального агенеза, синдром сакральной регрессии.
Определение и общие сведения
Синдром каудальной регрессии является редким врожденным пороком развития нижних сегментов позвоночника, включая аплазию или гипоплазию крестца и поясничного отдела позвоночника.
Распространенности оценивается примерно 1/50000 — 1/100000 беременностей. Диабет у матери является основным фактором риска для развития этого синдрома (в 200 раз выше, чем в общей популяции).
Этиология и патогенез
Синдром каудальной регрессии является результатом нарушения развития мезодермы первых 4 недель беременности. Этиология многофакторная, так как диабет у матери, сосудистая гипоперфузия и генетическая предрасположенность (например, мутации гена VANGL1) может приводить к рахвитию синдрома.
Клинические проявления
Существует целый спектр аномалий крестцового отдела туловища, пороки развития варьируют от изолированного частичного недоразвития крестцово-копчикового отдела позвоночника, до более тяжелых уродств. Эти аномалии развития могут приводить к деформации таза (как правило, слияние крыльев подвздошных костей), аномалиям нижних конечностей (изогнутые колени, варусная деформация), а также к двигательному и неврологическому дефициту различной степени тяжести (спонтанная двигательная активность и ослабленные глубокие сухожильные рефлексы нижних конечностей).
Могут также возникать сопутствующие осложнения со стороны мочеполовой, желудочно-кишечной и дыхательных систем. Мочеполовые аномалии могут включать в себя односторонний или двусторонний почечный агенез, эктопию почек, слитые мочеточники, что часто приводят к обструкции мочевыводящих путей, нейрогенному мочевому пузырю, энурезу или пузырно-мочеточниковому рефлюксу. Желудочно-кишечные аномалии могут включать в себя неспособность контролировать движения кишечника (недержание, энкопрез) и неперфорированной анус. Часто наблюдаются врожденные аномалии сердца. Также сообщалось о случаях с Киари мальформацией типа I, голопросенцефалией, хронической гипертонией и расщелинами губы и неба.
Диагностика
В самых тяжелых случаях диагноз формулируется в ходе пренатального УЗИ в первом триместре беременности. Тяжесть заболевания определяется с помощью УЗИ и МРТ новорожденного.
Дифференциальный диагноз
Основной дифференциальный диагноз сиреномелия. Синдром каудальной регрессии также ассоциирован с синдромом VACTERL. Синдром Куррарино является одной из форм синдрома каудальной регрессии, который характеризуется классической триадой симтомов: пресакральные и ретросакральные тканевые массы, дефект сакральной кости и аноректальные мальформации, имеет аутосомно-доминантное наследование и вызывается мутацией или делецией гена HLXB9.
Лечение
Лечение требует мультидисциплинарного подхода нейрохирургов, урологов, нефрологов, физиотерапевтов и психологов. Хирургические вмешательства, такие как трансуретроуретростомия, кожная везиктомия, катетеризация и/или введения антихолинолитиков, как правило, требуется для лечения урологических заболеваний. Колостома выполняется для лечения неперфорированного ануса. В зависимости от тяжести синдрома, ортопедические вмешательства также могут потребоваться. Лечение только поддерживающее, так как первичная патология является необратимой.
Прогноз
Прогноз неблагоприятный. Ранняя неонатальная смерть в тяжелых случаях происходит от осложнений со стороны сердца, почек и дыхательного тракта. Выжившие младенцы обычно имеют нормальную психическую функцию.
Источники (ссылки)[править]
Детская хирургия [Электронный ресурс] : учебник / под ред. Ю. Ф. Исакова, А. Ю. Разумовского; отв. ред. А. Ф. Дронов. — М. : ГЭОТАР-Медиа, 2015. — https://www.rosmedlib.ru/book/ISBN9785970434970.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Синдром каудальной регрессии (сакральная или люмбосакральная агенезия, синдром каудальной дисплазии, каудальная дисгенезия, сиреномелия) — редкий врожденный порок развития дистального отдела позвоночника и спинного мозга. Впервые врожденный дефект в виде агенезии дистальной части позвоночника описан в 1852 году.
24849 •
•
21.02.2019
Иллюстрация Георгия Сапего
В целом, синдром каудальной регрессии – широкое понятие, которое относится к гетерогенной группе врожденных каудальных аномалий, затрагивающих хвостовой отдел позвоночника и спинной мозг, заднюю кишку, мочеполовую систему и нижние конечности.
До настоящего времени точные причины возникновения синдрома каудальной регрессии неизвестны. Как правило, большинство случаев являются спорадическими. Предполагается, что это мультифакториальный порок, т.е. в его возникновении могут принимать участие много факторов: как генетическая предрасположенность, так и средовые факторы. Возможно, что порок может возникать в результате нарушения кровообращения нижней половины тела плода. Одним из наиболее достоверных факторов риска является сахарный диабет матери. Примерно от 15 до 25% матерей детей с каудальной дисгенезией страдают инсулинозависимым сахарным диабетом. Есть указания на участие в развитии каудальной регрессии гена VANGL1. Этот ген имеет большое значение в раннем развитии эмбриона.
Частота каудальной регрессии оценивается как 1 – 5 случаев на 100000 рождений.
Клинические проявления синдрома отличаются выраженной вариабельностью. В результате выделяются различные типы нарушений каудального отдела. В некоторых случаях у пациентов может быть только частичная агенезия крестцового отдела. В других случаях крестец может отсутствовать полностью. Агенезия крестцового отдела сопровождается недоразвитием бедер, гипоплазией ягодичных мышц, наличием ямки в нижней части спины (крестцовая ямка или крестцовый синус). В более тяжелых случаях в процесс вовлекаются и поясничные позвонки. Чем выше поражение, тем тяжелее клинические проявления.
Аномальное развитие каудальной области позвоночника может вызвать дополнительные нарушения, затрагивающие спинной мозг и нижние конечности. В некоторых случаях может произойти нарушение или повреждение нижней части спинного мозга, вызывающее различные неврологические нарушения, включая нарушение функции мочевого пузыря и кишечника, увеличение частоты мочеиспускания и невозможность полного опустошения мочевого пузыря (нейрогенный мочевой пузырь).
Повреждение нервов также может привести к аномалиям нижних конечностей. Такие нарушения могут включать сгибательные контрактуры колена и бедра. Контрактура — это состояние, при котором сустав становится постоянно неподвижным в согнутом или выпрямленном положении, полностью или частично ограничивая движение пораженного сустава.
Возможен широкий спектр дополнительных симптомов, включая аномалии почек, аномалии верхних позвонков, аномалии лица (расщелины губы и/или неба) и атрезии аноректального отдела.
В зависимости от степени тяжести поражения выделяют несколько типов заболевания. В 2002 г. была предложена новая классификация, связывавшая клинико-рентгенологический тип порока с потенциальной возможностью самостоятельной ходьбы.
тип А — небольшой дефект между подвздошными костями или сращение подвздошных костей по средней линии; отсутствие одного или нескольких поясничных позвонков;
тип B — полное сращение подвздошных костей, отсутствие нескольких поясничных позвонков;
тип С — полное сращение подвздошных костей между собой, отсутствие всех поясничных позвонков, значительный дефект между интактным грудным позвонком и тазом.
Самая тяжелая форма каудальной регрессии называется сиреномелией, или синдромом русалки. Однако некоторые авторы отмечают, что в литературе нет единого мнения относительно связи между синдромом каудальной регрессии и сиреномелией. В частности, есть мнение, что сиреномелия и синдром каудальной регрессии являются частью патогенетического спектра, обусловленного первичным дефицитом каудальной эмбриональной мезодермы.
Диагноз синдрома каудальной регрессии ставится, как правило, еще пренатально по результатам УЗИ, в остальных случаях он ставится почти сразу после рождения. После того, как выполнено УЗИ и диагноз подтвержден, новорожденных направляют на магнитно-резонансную томографию (МРТ) для определения тяжести случая.
Лечение симптоматическое и зависит от того, насколько серьезны проявления. В некоторых случаях ребенку могут понадобиться только специальная обувь, опоры для ног или костыли. Кроме того, рекомендуется лечебная физкультура. В более тяжелых случаях часто требуется хирургическое лечение. Как правило, лечение в таких случаях требует координированных усилий различных специалистов: педиатров, нейрохирургов, урологов, нефрологов, ортопедов. Прогноз зависит от тяжести заболевания.
Источник
из личного архива
Продолжение. Часть 1.
Тяжело писать. Имею ли я право разглашать диагнозы? Понятно, что мой сын даст мне любые разрешения, он мне доверяет. Мне кажется, об этом надо написать по двум соображениям — такие дети продолжают рождаться. И они очень пугают своих родителей. И, возможно, найдя эту статью, кто-то даст своему ребенку шанс вырасти в семье. Возможно. Ведь некоторые люди отказываются от своих детей только от страха. А могли бы прожить совсем другую жизнь, не расставаясь с ними.
Вот и Ромка ужасно напугал свою мать, когда у нее родился горбатый головастик с ластами тюленя вместо ног. Мне отдали его первый рентген, там именно такой силуэт. Кажется, акушерку он тоже перепугал, потому что его уронили и даже сломали руку. Врачи осмотрели и вздохнули. Объяснить маме, как все плохо, а будет еще хуже, это не та часть работы, ради которой становишься врачом. Мама попросила «отказную».
Вот ради этого момента тотального дна, когда новорожденный еще комочек остался один со своей огромной бедой, когда он, ни в чем не виноватый, уже лишился самого дорогого на земле, мамы, я хочу это все написать. Мы все попадаем в сложные ситуации, но все же мы не так беспомощны, как этот малыш. Мы должны верить в себя и верить в своих детей. Мы можем дать им шанс. Преодолеть свой страх.
Ромчик везунчик, мама отказалась от него по всем правилам и он через месяц уже поступил в дом малютки. Может, там не самые любящие руки, но они там есть. А это большое дело, мальчик начал развиваться.
85% детей с этим синдромом получают гидроцефалию, не могут ходить, не контролируют туалет, имеют проблемы с почками и другими внутренними органами.
Голова, действительно, была большая, но Ромчик обошелся без гидроцефалии, не знаю как. Никто не знает. Голова была большая по той причине, что тело сильно изуродовано, не хватало части позвоночника, не было бедер и самой попы в том виде, в котором мы к ним привыкли. Если бы не горбик, памперс было бы просто не за что зацепить. Большая голова была самой природой » рассчитана» на большое и крупное тело, а примерно до пояса Рома был крупным и крепким пацанчиком, поэтому только казалось, что она слишком большая. Это тело было маленьким. Крошечные ножки-кораблики. Было понятно, почему дети с этим синдромом не ходят.
А голова-то была еще и умной. С одной стороны, если ты мамин сын, это твой билет в хорошее будущее. А если ты сирота, то это твой постоянный кошмар. Ты все видишь, все понимаешь, но ничего не можешь сделать. Тебе постоянно очень и очень страшно. Ты один, настолько один, что начинаешь подвывать. Всю твою маленькую жизнь тебя терзают страхи, тревоги и беспокойства. Тебе катастрофически нужна мама, больше всего на свете тебе нужна мама, а ты не понимаешь, где она и почему не приходит. После «отказной» можно приходить. На самом деле в РФ нет никакой законной возможности отказаться от своего ребенка, разве что подложить его в беби-бокс. Родители, которые не хотят забирать младенца, подписывают «согласие на усыновление». Ну и , пока никто не усыновил, они имеют полное право навещать своих детей.
Мы приехали к Ромчику — и, знаете, хороший знак, он не особо к нам и пошел. То есть он не надеялся на всех незнакомых, как это бывает у детей с нарушением привязанности. Скорее, он нас побаивался. Врачи зачитали нам список диагнозов, который был совершенно не полный, в этой глуши не было нужного медицинского оборудования, и его первоначальный диагноз звучал как «много всяких нарушений». Правда, много. Прям на одного мальчишку чересчур много. Пока читали, он заснул у меня на руках. Трогательный, нежный, беззащитный.
«Ну и, конечно, ходить он никогда не будет» — сказали мне приговор. Ну, это было очевидно. Ходить ему было бы просто не на чем. Хорошо, что сам он об этом не знал. И да, мне было очень страшно.
Итак, первое свидание Рома проспал, второе он недоверчиво смотрел и слушал. На третьем он подарил мне все свое сердце и сказал «МАМА!» Бывает такая любовь, она нас сопровождает всю совместную жизнь. Он отдал мне все свое сердце, даже папе там почти не осталось места. И я всегда знаю, что он думает, чувствует, мне трудно поверить, что он не мой биологический сын.
«Ты любимый мамин сын!» — сказала ему я -«Манин син» — согласился он. И поехал с нами домой. Он вообще был очень отважный, как только получил поддержку в виду мамы, его отпустил страх. Он стал развиваться еще быстрее. И да, я хотела такого ребенка. На его инвалидность я почти не обращала внимания, все, что можно было бы сделать, мы бы с мужем сделали.
Единственное, чего я тогда не подумала, это что сделать-то ничего особо нельзя. Что это неоперируемый синдром и что даже просто вырасти, просто остаться живым это нередко невозможная задача. Потеряв одного ребенка, я совершенно не подумала, что со мной будет, если я потеряю и второго.
Продолжение . Все фото подлинные.
Источник
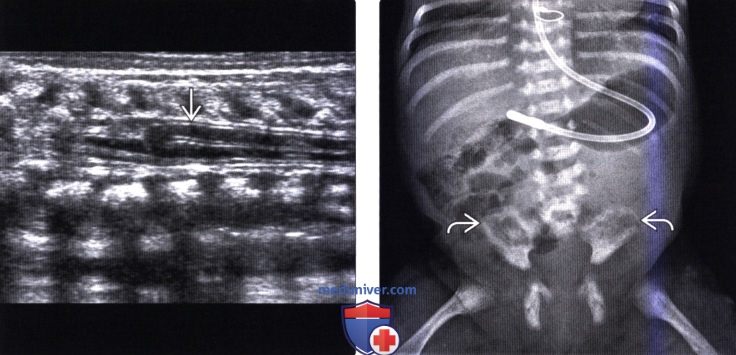
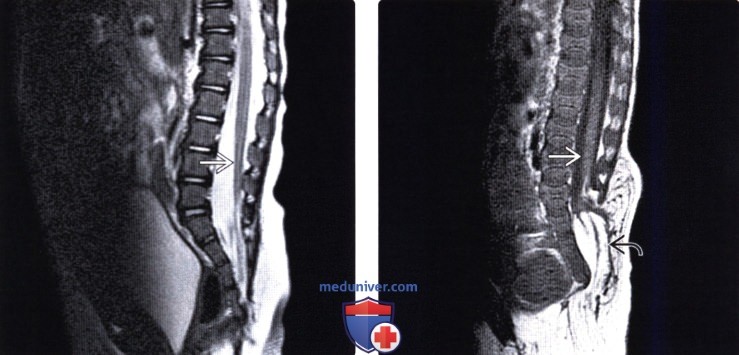
Лучевая диагностика синдрома каудальной регрессииа) Терминология: б) Визуализация: 1. Общие характеристики синдрома каудальной регрессии: 2. Рентгенологические данные: 3. КТ признаки синдрома каудальной регрессии: 4. МРТ признаки синдрома каудальной регрессии: 5. Ультразвуковые данные: 6. Несосудистые рентгенологические исследования: 7. Рекомендации по визуализации:
в) Дифференциальная диагностика синдрома каудальной регрессии: 1. Фиксированный спинной мозг: 2. Закрытая дизрафия позвоночника: 3. Скрытое крестцовое менингоцеле (СКМ): г) Патология: 1. Общие характеристики синдрома каудальной регрессии: 2. Стадирование, степени и классификация: 3. Макроскопические и хирургические особенности: д) Клинические особенности: 1. Клиническая картина синдрома каудальной регрессии: 2. Демография: 3. Течение заболевания и прогноз: 4. Лечение: е) Диагностическая памятка: ж) Список использованной литературы: — Также рекомендуем «МРТ при терминальном миелоцистоцеле (сирингоцеле)» Редактор: Искандер Милевски. Дата публикации: 18.7.2019 |
Источник