Синдром эдвардса код по мкб

Рубрика МКБ-10: Q91.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q90-Q99 Хромосомные аномалии, не классифицированные в других рубриках / Q91 Синдром Эдвардса и синдром Патау
Определение и общие сведения[править]
Cиндром Эдвардса
Синонимы: трисомия 18
Трисомия по 18-й хромосоме встречается у новорожденных с частотой от 1:3300 до 1:10 000; у девочек бывает в 3 раза чаще, чем у мальчиков. Больные дети часто рождаются недоношенными или переношенными. Нарушения при трисомии по 18-й хромосоме гораздо тяжелее, чем при синдроме Дауна; лишь 50% пробандов доживают до 2-месячного возраста; 10% живут 1 год. Средняя продолжительность жизни мальчиков — 60, девочек — 280 дней.
Этиология и патогенез[править]
Большинство случаев связаны со свободной трисомией 18. Мозаичная трисомия 18 была обнаружена у нескольких пациентов с клинической картиной, которая варьируется от классической трисомии 18 до нормального фенотипа в зависимости от количества трисомных клеток, присутствующих в тканях. Фенотип трисомии 18, по-видимому, связан с наличием трех копий интервала 18q11-q12.
Клинические проявления[править]
Клиническая картина: череп необычной формы (узкий лоб и широкий выступающий затылок), низкое расположение ушей, микрогнатия, сгибательная контрактура кистей и стоп, дисплазия стоп, пороки сердца, сильная задержка психического развития. Главные нарушения обмена веществ и эндокринные расстройства: гипоплазия подкожной клетчатки, сильная задержка роста. Дисгенезия щитовидной железы или надпочечников встречается менее чем у 10% больных.
Синдром Эдвардса неуточненный: Диагностика[править]
Трисомию 18 можно заподозрить во время беременности по результатам ультразвукового исследования плода (задержка роста, пороки развития, множественные кисты сосудистого сплетения) и подтверждить кариотипическим анализом плода. Маркеры сыворотки (используемые для диагностики трисомии 21) также могут быть аномальными.
Дифференциальный диагноз[править]
Синдром Эдвардса неуточненный: Лечение[править]
Хирургическое лечение пороков развития мало способствует улучшению неблагоприятного прогноза, связанного с этим синдромом: 90% детей умирают в течение первого года жизни от сердечных, почечных или неврологических осложнений или от повторных инфекций.
Сообщалось о длительном выживании (в некоторых случаях до взрослого возраста), главным образом в случаях мозаичной или частичной трисомии (в результате транслокации). Большинство немозаичных пациентов имеют лишь ограниченную автономию (отсутствие речи и ходьбы).
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
1. Buyse ML. Birth Defects Encyclopedia. Cambridge: Blackwell, 1990.
2. Emery A, Rimoin D. Principles and Practice of Medical Genetics (2nd ed). New York: Churchill Livingstone, 1990.
3. Gorlin RJ, et al. Syndromes of the Head and Neck (3rd ed). New York: Oxford University Press, 1990.
4. Hall JG, et al. Handbook of Normal Physical Measurements. New York: Oxford University Press, 1989.
5. Jones KL. Smith’s Recognizable Patterns of Human Malformation (4th ed). In M Markowitz (ed.), Major Problems in Clinical Pediatrics (vol VII). Philadelphia: Saunders, 1988.
6. McKusick V. Mendelian Inheritance in Man (10th ed.). Baltimore: Johns Hopkins University Press, 1992.
7. Rimoin DL. Disorders of the Endocrine Glands. In AA Dietz (ed.), Genetic Disease: Diagnosis and Treatment. Proceedings of the Fifth Arnold O. Beckman Conference in Clinical Chemistry. Monterey, CA: The Association for Clinical Chemistry, 1983.
8. Taybi H, Lachman RS. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias (3rd ed). Chicago: Yearbook, 1990.
9. Vogel F, Motulsky AG. Human Genetics: Problems and Approaches (2nd ed). New York: Springer, 1986.
10. Wiedmann H-R, et al. Atlas of Clinical Syndromes—A Visual Aid to Diagnosis (2nd ed). St. Louis: Mosby, 1989.
11. Wynne-Davis R, Hall CM, Apley AG. Atlas of Skeletal Dysplasias. Edinburgh: Churchill Livingstone, 1985.
Действующие вещества[править]
Источник
Содержание
- Описание
- Симптомы
- Диагностика
- Лечение
Названия
Синдром Эдвардса.
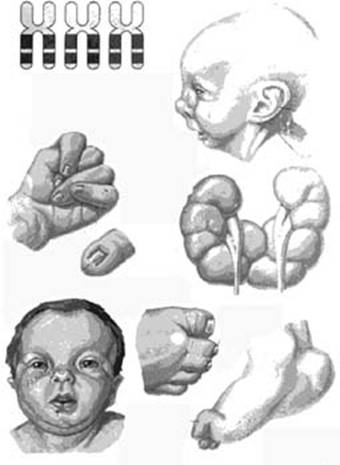
Симптомы при синдроме Эдвардса (трисомия 18)
Описание
Впервые трисомию по группе Е описали J. Edwads и соавт. (1960г. ). Частота синдрома Эдвардса среди новорожденных в среднем составляет 1:7000, замечено, что девочки поражаются в 3 раза чаще, чем мальчики. Ученым высказано предположение о стабилизирующем действии Х-хромосомы при абберациях пары 18, тогда как зиготы с трисомией 18, имеющие мужской генотип, элиминируются. Средний возраст матерей 32,5 года, отцов – 35 лет.
Симптомы
Длительность беременности превышает нормальную, и составляет в среднем 42 недели. Во время беременности диагностируют слабую активность плода и многоводие. Отмечается,что плацента имеет меньшие, чем обычно, размеры, часто обнаруживается только одна пупочная артерия; многие дети рождаются в асфиксии, с очень низкой массой тела и выраженной гипотрофией.
Фенотип больных с синдромом Эдвардса довольно характерен. Череп долихоцефалический, с низким лбом, затылок более широкий и выступающий. Часто встречается микроцефалия или гидроцефалия. Надглазничные валики сглажены, глазные щели узкие, наблюдается эпикант, птоз, часто встречается глазная патология, микрофтальмия, колобома, катаракта. Переносье вдавлено, но спинка носа тонкая, выступающая, ушные раковины расположены очень низко, диагностируется отсутствие мочки и козелка, недоразвиты завиток и противозавиток. Характерна микроретрогнатия. Рот мешьше обычных размеров, имеет треугольную форму, верхняя губа короче обычного. Небо высокое, иногда с расщелиной, шея короткая, нередко с крыловидной складкой.
Аномалии опорно-двигательного аппарата отличаются разнообразием: расширение шрудной клетки, укорочение грудины, таз узкий, деформации конечностей, ограничена подвижность в тазобедренных суставах, описаны вывихи бедра. Кисти и пальцы короткие, клинодактилия 5 пальцев кисти, дистально расположенный, гипоплазированный 1 палец кисти, сглажен тенар. Пальцы сжаты в кулак по типу «флексорной аномалии»: 2 и 5 пальцы расположены сверху и прикрывают прижатые к ладони 3 и 4 пальцы; 1 палец стопы широкий и короткий, синдактилия 2 и 3 пальцев. Типична для трисомии 18 форма стопы в виде своеобразной «качалки». Характерна общая мышечная гипотония. У мальчиков нередко встречается крипторхизм, гипоспадия; гипертрофия клитора у девочек.
Интеллектуальный дефект соответствует олигофрении в степени идиотии ил глубокой имбецильности и только в единичных описаниях мозаичного варианта трисомии 18 умственное недоразвитие менее грубое. Нередко у больного развиваются судороги.
Внешний вид больного с синдромом Эдвардса (трисомия 18)
Диагностика
Дерматографическая картина при синдроме Эдвардса отличается своеобразными признаками: отмечается большая частота дуг на подушечках пальцев рук (примерно в 10 раз выше, чем в общей популяции), нередко дистальная сгибательная складка на пальцах не развита, у трети больных обнаруживается поперечная ладонная борозда, гребневый счет увеличен, осевой трирадиус обычно расположен дистально.
При аутопсии при синдроме Эдвардса находят разнообразные пороки развития практически всех органов и систем. С различной частотой встречаются аномалии ЦНС: аплазия или гипоплазия мозолистого тела, гипоплазия мозжечка, атрофия мозговых извилин. У 95% пациентов с трисомией 18 диагностируются пороки сердца и крупных сосудов, наиболее часто встречаются дефект межжелудочковой перегородки и незаращение артериального протока. Около половины всех случаев синдрома Эдвардса сопровождается аномалиями органов пищеварения: нарушение расположения кишечника, меккелев дивертикул, атрезия пищевода, атрезия заднего прохода. В 50% случаев отмечаются пороки развития мочеполовой системы – дольчатая или подковообразная почка, дупликация мочеточников, гипоплазия яичников.
При цитогенетическом обследовании в 80% случаев находят трисомию 18, у 10% больных – мозайцизм. Описаны случаи транслокационного варианта, двойной анеуплодии типа 48, ХХУ +18 с участием трисомного по хромосоме 18 клона.
Лечение
Нет патогенетического лечения синдрома Эдвардса на сегодняшний день, возможна лишь симптоматическая коррекция патологий. Прогноз для жизни неблагоприятный, средняя продолжительность жизни мальчиков 2-3 месяца, девочек – 10 месяцев. 30% больных умирают в течение 1-го года жизни, до года доживает лишь 10% больных. При мозаичных вариантах прогноз для жизни несколько лучше.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 мая 2015;
проверки требуют 16 правок.
Синдром Э́двардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом (John H. Edwards). Популяционная частота примерно 1:3000 в США, и 1:5000 в мире на 2016 год. Дети с трисомией в 18 хромосоме чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21[3] и 13[4]. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. Выживание после года жизни составляет около 5–10%[5].
Причины заболевания[править | править код]
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома[править | править код]
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Прогноз[править | править код]
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления[править | править код]
Частота появления синдрома Эдвардса составляет ~ 1:7000 зачатий и 1:8000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Вариации[править | править код]
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикреплённая к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
См. также[править | править код]
- Анеуплоидия
- Хромосомные болезни
- Синдром Дауна
- Синдром Патау
Примечания[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ Синдром Дауна
- ↑ Синдром Патау
- ↑ Genetics Home Reference. Trisomy 18 (англ.). Genetics Home Reference. Дата обращения 20 сентября 2019.
Ссылки[править | править код]
- https://rh-conflict.narod.ru/student/lectures/hrombol.htm
Источник
Q90 Синдром Дауна
- Q90.0 Трисомия 21, мейотическое нерасхождение
- Q90.1 Трисомия 21, мозаицизм митотическое нерасхождение
- Q90.2 Трисомия 21, транслокация
- Q90.9 Синдром Дауна неуточненный
Q91 Синдром Эдвардса и синдром Патау
- Q91.0 Трисомия 18, мейотическое нерасхождение
- Q91.1 Трисомия 18, мозаицизм митотическое нерасхождение
- Q91.2 Трисомия 18, транслокация
- Q91.3 Синдром Эдвардса неуточненный
- Q91.4 Трисомия 13, мейотическое нерасхождение
- Q91.5 Трисомия 13, мозаицизм митотическое нерасхождение
- Q91.6 Трисомия 13, транслокация
- Q91.7 Синдром Патау неуточненный
Q92 Другие трисомии и частичные трисомии аутосом, не классифицированные в других рубриках
- Q92.0 Полная хромосомная трисомия, мейотическое нерасхождение
- Q92.1 Полная хромосомная трисомия, мозаицизм митотическое нерасхождение
- Q92.2 Большая частичная трисомия
- Q92.3 Малая частичная трисомия
- Q92.4 Удвоения, наблюдаемые только в прометафазе
- Q92.5 Удвоения с другим комплексом перестроек
- Q92.6 Особо отмеченные хромосомы
- Q92.7 Триплоидия и полиплоидия
- Q92.8 Другие уточненные трисомии и частичные трисомии аутосом
- Q92.9 Трисомии и частичные трисомии аутосом неуточненные
Q93 Моносомии и утраты части аутосом, не классифицированные в других рубриках
- Q93.0 Полная хромосомная моносомия, мейотическое нерасхождение
- Q93.1 Полная хромосомная моносомия, мозаицизм митотическое нерасхождение
- Q93.2 Хромосомное смещение с закруглением или смещением центра
- Q93.3 Делеция короткого плеча хромосомы 4
- Q93.4 Делеция короткого плеча хромосомы 5
- Q93.5 Другие делеции части хромосомы
- Q93.6 Делеции, наблюдаемые только в прометафазе
- Q93.7 Делеции с другим комплексом перестроек
- Q93.8 Другие делеции из аутосом
- Q93.9 Делеция из аутосом неуточненная
Q95 Сбалансированные перестройки и структурные маркеры, не классифицированные в других рубриках
- Q95.0 Сбалансированные транслокации и инсерции у нормального индивида
- Q95.1 Хромосомные инверсии у нормального индивида
- Q95.2 Сбалансированные аутосомные перестройки у нормального индивида
- Q95.3 Сбалансированные половые/аутосомные перестройки у нормального индивида
- Q95.4 Индивиды с обозначенным гетерохроматином
- Q95.5 Индивиды с ломким участком аутосом
- Q95.8 Другие сбалансированные перестройки и структурные маркеры
- Q95.9 Сбалансированные перестройки и структурные маркеры неуточненные
Q96 Синдром Тернера
- Q96.0 Кариотип 45,Х
- Q96.1 Кариотип 46,Х iso Xq
- Q96.2 Кариотип 46,Х с аномальной половой хромосомой, за исключением iso Xq
- Q96.3 Мозациизм, 45,Х/46,ХХ или ХУ
- Q96.4 Мозациизм, 45,Х/другая клеточная линия линии с аномальной половой хромосомой
- Q96.8 Другие варианты синдрома Тернера
- Q96.9 Синдром Тернера неуточненный
Q97 Другие аномалии половых хромосом, женский фенотип, не классифицированные в других рубриках
- Q97.0 Кариотип 47,ХХХ
- Q97.1 Женщина с более чем тремя Х-хромосомами
- Q97.2 Мозациизм, цепочки с различным числом Х-хромосом
- Q97.3 Женщина с 46,ХУ-кариотипом
- Q97.8 Другие уточненные аномальные половые хромосомы, женский фенотип
- Q97.9 Аномалия половых хромосом, женский фенотип, неуточненная
Q98 Другие аномалии половых хромосом, мужской фенотип, не классифицированные в других рубриках
- Q98.0 Синдром Клайнфелтера, кариотип 47,ХХУ
- Q98.1 Синдром Клайнфелтера, мужчина с более чем двумя Х-хромосомами
- Q98.2 Синдром Клайнфелтера, мужчина с 46,ХХ-кариотипом
- Q98.3 Другой мужчина с 46,ХХ-кариотипом
- Q98.4 Синдром Клайнфелтера неуточненый
- Q98.5 Кариотип 47,ХУУ
- Q98.6 Мужчина со структурно измененными половыми хромосомами
- Q98.7 Мужчина с мозаичными половыми хромосомами
- Q98.8 Другие уточненные аномалии половых хромосом, мужской фенотип
- Q98.9 Аномалия половых хромосом, мужской фенотип, неуточненная
Q99 Другие аномалии хромосом, не классифицированные в других рубриках
- Q99.0 Мозаик [химера] 46,ХХ/46,ХУ
- Q99.1 46,ХХ истинный гермафродит
- Q99.2 Ломкая Х-хромосома
- Q99.8 Другие уточненные хромосомные аномалии
- Q99.9 Хромосомная аномалия неуточненная
Источник
Все материалы
Клинические протоколы МЗ РК
ҚР ДСӘДМ клиникалық хаттамалар
Клинические рекомендации МЗ РФ
Клинические протоколы МЗ РБ
Международные клинические руководства
Обзорные статьи по заболеваниям
- Ещё
Клинические протоколы МЗ РК — 2020
Клинические протоколы МЗ РК — 2019
Клинические протоколы МЗ РК — 2018
Клинические протоколы МЗ РК — 2017
ҚР ДСӘДМ клиникалық хаттамалар — 2017
Клинические протоколы МЗ РК — 2016
Клинические протоколы МЗ РК — 2015
Клинические протоколы МЗ РК — 2014
ҚР ДСӘДМ клиникалық хаттамалар — 2014
Клинические протоколы МЗ РК — 2013
Архив — Клинические протоколы МЗ РК — 2012 (Приказы №883, №165)
Архив — Клинические протоколы МЗ РК (Протокол №8 от 17.04.2012 г., Экспертный совет МЗ РК)
Архив — Клинические протоколы МЗ РК — 2010 (Приказ №239)
Архив — Клинические протоколы МЗ РК — 2007 (Приказ №764)
Архив — Аурулардың диагностикасы және емдеу хаттамалары (Приказ №764, 2007, №165, 2012)
Архив — Протоколы диагностики и лечения Министерства здравоохранения Республики Казахстан (2006, устар.)
Клинические протоколы (Беларусь)
Клинические рекомендации РФ (Россия) 2013-2017
Клинические рекомендации РФ (Россия) 2018-2020
Международные клинические руководства для использования в РК
Источник