Синдром гемифациальной микросомии синдром гольденхара

Гемифациальная микросомия (синонимы: синдром первой и второй жаберных дуг, синдром Гольденхара, краниофациальная микросомия, отомандибулярный дизостоз и латеральная фациальная дисплазия) является редким наследственным заболеванием, характеризующимся значительным числом аномалий, которые возникают вследствие нарушения развития первой и второй жаберных дуг первого глоточного кармана, первой жаберной щели и зачатков височной кости. Среди врожденных пороков развития черепно-челюстно-лицевой области гемифациальная микросомия занимает второе место по частоте встречаемости после врожденных расщелин верхней губы и нёба. Частота этого синдрома колеблется в пределах 1:3500-5600 новорожденных, он присутствует у 1 из 1000 у детей с врожденной глухотой. Распределение по половому признаку (мужчины и женщины) составляет примерно 3:2.
Этиология и тип наследования изучены недостаточно. Неблагоприятный акушерско-гинекологический анамнез матери (предшествующие аборты, сахарный диабет, избыточный вес) и тератогенные факторы на ранних сроках беременности являются отягощающими факторами риска рождения больного ребенка. Вероятность повторного рождения больного ребенка, как и вероятность рождения больного ребенка у носителя патологии, ориентировочно равна 2%.
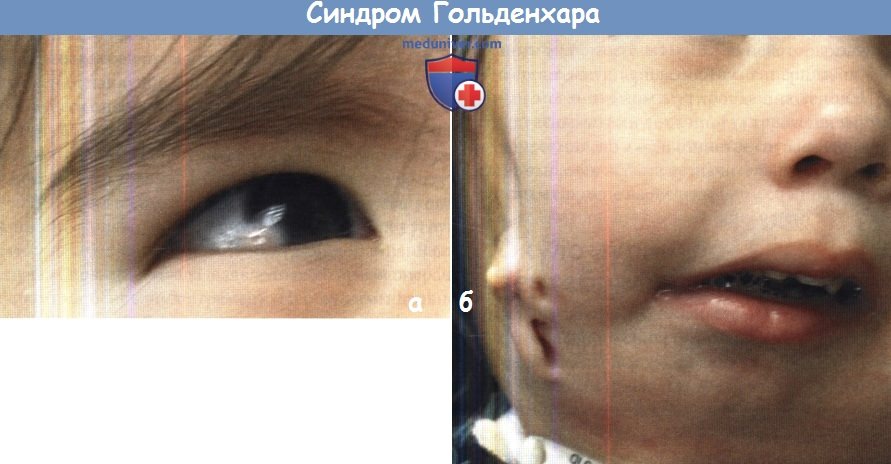
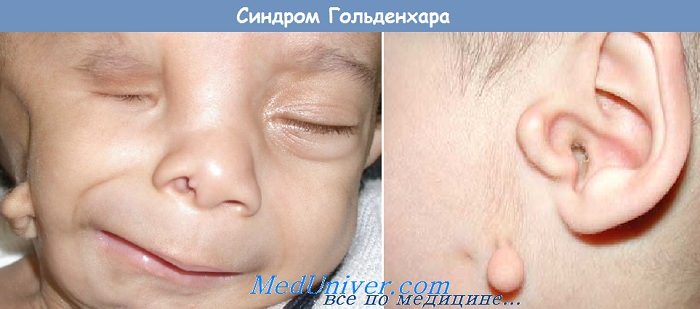
Клинически для гемифациальной микросомии характерны: недоразвитие тела и ветви нижней челюсти, гипоплазия скуловой кости и дуги, недоразвитие структур ВНЧС; аплазия ветви нижней челюсти и ВНЧС; нарушение размеров и положения глазницы; гипоплазия, аплазия ушной раковины, атрезия слухового прохода, поражение лицевого нерва, гипоплазия мимических мышц; дефицит мягких тканей; макростомия; предушные придатки и свищи; иногда сочетание с врожденной расщелиной губы и неба, эпибульбарным дермоидом, аномалией прикуса, адентией, нарушением структуры эмали и формы зубов, пороками развития опорно-двигательного аппарата, органов зрения и нервной системы, а также аномалиями мочевыделительной системы и желудочно-кишечного тракта.
В связи с малой информированностью населения России о врожденной патологии, необходимо как можно раньше получить консультацию челюстно-лицевого хирурга и ортодонта, в качестве необходимого обследования рекомендуется компьютерная томография черепа. Всем больным необходимо проверить остроту слуха.
Таким образом, ввиду многообразия клинических признаков, становится ясным, что лечение таких пациентов носит мультидисциплинарный подход. Лечение многоэтапное и зависит от вида (объема) патологии, комбинации различных нарушений и возраста пациента. В легких случаях можно ограничиться наблюдением до возраста 2-3 лет. Затем, по показаниям, начинать ортодонтическое и хирургическое лечение. В сложных случаях (наличие дефекта челюсти, расщелины неба, микросомии) хирургическое лечение проводится в возрасте до 1 или 2-х лет. Затем рекомендуется комплексное лечение (ортодонтическое, хирургическое, логопедическое и т.д.)
Хирургические этапы лечения включают: применение компрессионно-дистракционного остеогенеза, эндопротезирование ВНЧС, реконструктивные операции: остеотомия верхней и нежней челюсти, пластические операции.
Ортодонтические этапы лечения направлены: на предупреждения развития асимметрии нижней челюсти, устранение аномалий прикуса, подготовки зубных рядов, жевательных и мимических мышц для хирургических этапов лечения. Создание множественных окклюзионных контактов.
Ортодонтическое лечение можно разделить на 3 этапа
Этап 1
Период временного (молочного) прикуса.
Пожалуй, это самый важный период, ведь главными задачами в это время являются информирование родителей о пороке развития, тяжести и возможных дальнейших рисках в развитии заболевания. Родителям следует рассказать о правилах гигиены полости рта пациента.
С возрастом, если ребенок не получает квалифицированного лечения, недоразвитие костей лицевого скелета прогрессирует. Патология прикуса становится более выраженной, затрудняется жевание и глотание, нарастает деформации, нарастает асимметрия, что в свою очередь ведет к психологической травме ребенка и его родителей.
Начало ортодонтического лечения предусматривает применение съемных эластичных двучелюстных аппаратов, съёмных индивидуальных пластинчатых аппаратов и съёмных индивидуальных одночелюстных пластинчато-каповых аппаратов.
Целью ортодонтического лечения является знакомство и мягкая адаптация пациента к ортодонтическим аппаратам, применение которых направлено на устранение асимметрии нижней челюсти, а в случае отсутствия или минимально выраженной асимметрии — предупреждение ее развития.
Этап 2
Период сменного прикуса
Задачами этого периода ортодонтического лечения являются устранение аномалий прикуса, размеров и формы зубоальвеолярных дуг. По прежнему остаются актуальными задачи предупреждения и устранения асимметрии нижней челюсти в связи с ее патологическим односторонним недоразвитием.
Лечение чаще проводится съёмными пластинчатыми расширяющими опорно-удерживающими аппаратами. Также используются несъёмные индивидуальные каркасные аппараты.
Этап 3
Период позднего сменного и постоянного прикуса.
На этом этапе продолжается ортодонтическое лечение, предусматривающее дальнейшее устранение патологий прикуса, подготовку зубных рядов для окончательных этапов хирургического лечения, создание множественных окклюзионных контактов.
Лечение проводится с использованием несъемной ортодонтической аппаратуры – брекет системы.
Заключительным этапом ортодонтического лечения является ретенционный период. Целью данного периода являются закрепление и удержание результатов ранее проведенного лечения. Используются несъемные проволочные ретейнеры, которые фиксируются на нёбную (язычную) поверхность передней группы зубов, что делает их “невидимыми” для окружающих. Совместно с несъёмными ретейнерами в зубочелюстной лаборатории изготавливается съемный индивидуальнымй двучелюстной ретенционный аппарат, который ребенок использует во время сна.
Ретенционный период продолжается до полного окончания роста ребенка. Рекомендуется время от времени проходить осмотр у челюстно-лицевого хирурга и ортодонта для предупреждения непредвиденных ситуаций, ведь одним из важнейших факторов в лечении такой сложной врождённой патологии является заинтересованность родителей и выполнение рекомендаций врачей.
После ортодонтического лечения (или в его процессе) может быть необходима хирургическая коррекция (пластика ушной раковины, контурная пластика нижней челюсти и т.д.)
Реабилитация пациента должна быть закончена к 16-18 годам.
Источник

Синдром Гольденхара — наследственная односторонняя гипоплазия лица с преимущественным поражением ушной раковины, глаз, носа, зубов, позвоночника. При этом вторая половина лица имеет абсолютно нормальный вид. Это редко встречающееся, генетически обусловленное заболевание отличается неполноценным развитием скелета, мышц, сосудисто-нервных стволов и прочих структур человеческого организма.
Окуло-аурикуло-вертебральная дисплазия — второе название данного недуга. Синдром Гольденхара представляет собой отдельный вид целой группы патологий, для которой характерна задержка физического развития человека. Непосредственной причиной его развития является внутриутробное повреждение жаберных дуг, из которых у плода формируются структуры слухового и жевательного аппарата.
Синдром Гольденхара отличается широким клиническим полиморфизмом. Классическими симптомами заболевания являются: недоразвитие в процессе эмбриогенеза нижней челюсти, резкое нарушение симметрии лица, деформация наружного уха и глаза, нарушение строения позвонков шеи. Возможно полное отсутствие ушной раковины или органа зрения. У больных формируются дермоидные кисты в глазах, развиваются отклонения в работе сердечно-сосудистой, мочеполовой систем и желудочно-кишечного тракта. Клинически это проявляется одышкой, нарушением процесса питания, снижением слуха с пораженной стороны. Синдром Гольденхара не связан с умственной отсталостью. Выявить патологию можно на 20-24 неделе беременности с помощью ультразвуковой диагностики со сканированием в трех измерениях. Большинство случаев синдрома – спорадические.

Впервые патологию описал в 1952 году американский доктор Гольденхар. Синдром получил свое название по фамилии его первооткрывателя. У мальчиков заболевание встречается несколько чаще, чем у девочек. С помощью современных методов диагностики заболевание обнаруживают внутриутробно и решают вопрос о целесообразности сохранения беременности.
Этиология и патогенез
Причины возникновения и механизмы развития синдрома Гольденхара остаются неизвестными. Мнения ученых сходятся на том, что патология относится к наследственным недугам. У пациентов с данным синдромом обнаруживают хромосомные аномалии и мутации генов. Больной ребенок рождается с множественными дефектами лица и патологиями внутренних органов.

Факторы риска рождения больных детей:
- кровнородственный брак,
- частые аборты у матери,
- тяжелые эндокринопатии,
- лишний вес матери,
- воздействие химических веществ и патогенных биологических агентов на ранних сроках беременности.
Во время замены источника кровоснабжения на участке 1 и 2 жаберных щелей эмбриона происходит кровоизлияние. Разрыв кровеносного сосуда в этом месте приводит к нарушению митотического деления клеток и неправильному формированию основных анатомических структур человеческого организма.
Симптоматика
Первые симптомы патологии можно заметить сразу после рождения ребенка. У больных детей асимметричное лицо, мелкие глазницы, деформированные ушные раковины, недоразвитая нижняя челюсть. Это внешние признаки патологии, которые нередко сопровождаются патологическими изменениями во внутренних органах.

Симптомы со стороны зрительного анализатора:
- Анофтальмия,
- Микрофтальм,
- Микрокорнеа — малая роговица,
- Аниридия — отсутствие радужки,
- Птоз верхнего века,
- Страбизм – отклонение зрительной оси глаза от точки фиксации,
- Помутнение хрусталика,
- Дермоиды и липодермоиды на поверхности глазного яблока – небольшие опухоли,
- Поражение глазодвигательных мышц.
Клинические признаки при поражении ушей:
- Аномальное расположение ушных раковин,
- Минимальные размеры ушей,
- Деформация ушных раковин,
- Плоская или выступающая форма ушей,
- Характерные выросты на ушных раковинах,
- Дефекты барабанной перепонки,
- Соединение ушной раковины с уголком рта, придающее лицу перекошенный вид,
- Сужение слухового прохода,
- Фистулы и околоушные свищи,
- Отсутствие наружного слухового прохода,
- Снижение слуха,
- Двусторонняя асимметричная кондуктивная тугоухость,
- Полное отсутствие слуха.
Нижняя челюсть у больных недоразвита. Одна половина лица меньше другой, мимические мышцы развиты слабо. Небо имеет вид высокой арки, иногда расщепляется. Ротовая щель при этом слишком широкая, прикус нарушается, появляются добавочные уздечки и расщелины на малом язычке. Нарушается рост зубов. Мягкое небо, губы и гланды деформированы. Имеются трахеопищеводные свищи. Рот большой, один угол выше другого. Недоразвиты скулы, лоб выступает, присутствуют аномалии языка.

У деток с синдромом Гольденхара имеются проблемы с позвоночником: недоразвитие шейного отдела, сколиоз, косолапость, клиновидная форма позвонков, их слияние, наличие полупозвонков, сращение шейных позвонков с затылком, костные аномалии, короткая шея, искривление позвоночника.
Проявлениями со стороны сердечно-сосудистой системы являются симптомы врожденных пороков сердца. Больные дети рождаются с недоразвитием легких, отсутствием органов малого таза, парезом лицевого нерва, гидроцефалией. Они испытывают трудности в обучении. Каждый 10 больной ребенок рождается с поражением ЦНС.
Для синдрома Гольденхара характерно появление симптоматики с одной стороны лица и туловища.
Осложнения и последствия

Некоторые проявления синдрома Гольденхара не совместимы с жизнью. Ребенок может умереть сразу после рождения.
Осложнения синдрома Гольденхара:
- неправильный прикус вызывает ряд неудобств,
- развивается гипоплазия лицевых костей,
- трудности с глотанием и жеванием,
- прогрессирующая патология зрения,
- серьезные физические неудобства,
- психологическая травма.
Диагностика
Предварительный диагноз патологии устанавливают сразу после рождения ребенка на основании визуального осмотра. Диагностика включает тщательный опрос родителей и составление анамнеза.
Разнообразные диагностические процедуры позволяют поставить точный диагноз.

- Определение остроты слуха – регистрация активности слухового нерва после короткой акустической стимуляции, аудиометрия, импедансометрия.
- Осмотр глаза и проведение офтальмологического исследования.
- Электрокардиография.
- Ренгенография черепа, шеи, грудной клетки.
- УЗИ внутренних органов.
- КТ головного мозга.
- Медико-генетическое консультирование.
Лечение
Лечение синдрома Гольденхара мультидисциплинарное. Дети до трехлетнего возраста наблюдаются у различных специалистов — ЛОР-врача, окулиста, сурдолога, ортопеда и прочих. Детей старше 3 лет направляют к хирургу. В тяжелых случаях для удаления грубых врожденных дефектов сразу после рождения проводят оперативное лечение, а затем комплексную медикаментозную и ортодонтическую терапию.
Хирургическое лечение
Вид оперативного вмешательства определяется тяжестью патологии.
- После искусственного перелома кости соединяют костные фрагменты и устраняют их подвижность с помощью компрессионно-дистракционных аппаратов.
- Проводят протезирование и реконструкцию височно-нижнечелюстного сустава.
- Устраняют деформации носа и улучшают его функции путем искусственного перелома носовой кости.
- Исправляют патологический прикус оперативным путем.
- Проводят пластические операции — пластику носа, ушных раковин, нижней челюсти.

Чтобы предотвратить развитие воспалительных осложнений, во время реабилитации назначают антибиотики и витамины. К остеотропным антибактериальным препаратам относятся «Линкомицин», «Эритромицин», защищенные пенициллины – «Амоксиклав». Для снятия боли назначают анальгетики – «Нурофен», «Ибуклин». Больные дети должны полноценно питаться и принимать поливитаминные комплексы.
Физиотерапия – неотъемлемый компонент лечебного и реабилитационного процесса любых заболеваний. Больным с синдромом Гольденхара проводят УФО, ультразвук, магнитотерапию, лазерное лечение, оксигенотерапию. В терапевтический комплекс входит также лечебная гимнастика, занятия с сурдологом и психологом, слухопротезирование цифровыми имплантируемыми слуховыми аппаратами, контроль слуха и периодические настройки аппаратов. При сколиозе доктора назначают массаж и ношение специальных корсетов.
Лечение у ортодонта проходит в несколько этапов. Первый этап — молочный. Он представляет собой знакомство с патологией. Специалисты объясняют родителям, как правильно ухаживать за полостью рта больного ребенка, предупреждают о возможных осложнениях, проводят аппаратное лечение для исправления дефектов челюсти. Следующий этап — сменный, заключающийся в исправлении прикуса и коррекции имеющихся деформаций во рту. Постоянный этап – замена съемных аппаратов на брекеты и различные фиксаторы. Обычно к 18-летнему возрасту ортодонтическое лечение полностью завершается.
Народная медицина
Наиболее распространенные рецепты народной медицины:
- для промывания и очищения глаз можно использовать отвары и настои лекарственных трав – василька, подорожника, ромашки, тмина,
- употреблять внутрь средство, приготовленное из меда и сока калины,
- закапывать в уши настой из масла шиповника и семян аниса,
- принимать внутрь настой из корня аира.
Прогноз

Длительное, раннее и комплексное лечение заболевания в большинстве случаев делает прогноз благоприятным. Пластические операции в некоторых случаях полностью устраняют внешние признаки заболевания. Это позволяет детям посещать общественные места, учиться, а в дальнейшем – работать, полноценно жить, создавать семью и заводить детей. После проведения ряда операций ребенок становится совершенно здоровым.
Если ребенок имеет не только внешние признаки патологии, но и поражение внутренних органов, он получает инвалидность. В случаях с особо тяжелыми проявлениями прогноз неблагоприятен. Длительность жизни больных с синдромом Гольденхара зависит от тяжести поражения жизненно важных органов.
Профилактика
Специфической профилактики синдрома Гольденхара не существует. К общим профилактическим мерам, позволяющим предотвратить это врожденное заболевание, относятся:
- отсутствие хронических гинекологических заболеваний у матери,
- правильное питание будущей матери,
- отказ родителей от вредных привычек,
- оптимальная физическая активность,
- обоюдное желание иметь ребенка,
- взаимопонимание, теплая и любящая атмосфера в семье.
Пренатальная диагностика врожденных аномалий заключается в проведении фетоскопии и ультразвукового сканирования эмбриона. Проводят УЗИ на 20-24 неделе беременности. Этот метод является точным на 100%. Он позволяет специалистам понять, стоит ли сохранять беременность.
Источник
Сопутствующие болезни синдрома Гольденхара (окуло-аурикуло-вертебральной дисплазии) в отоларингологииГемифациальная микросомия является частью окуло-аурикуло-вертебрального синдрома. Наиболее часто встречающейся клинической формой является синдром Гольденхара. Основными признаками его являются пороки развития ушной раковины, которая обычно расположена на половине лица с микросомией, эпибульбарные дермоиды, пороки развития позвоночника, колобома верхнего века, преаурикулярная папиллома, агенезия околоушной слюнной железы с одной стороны, врожденный паралич лицевого нерва. Выраженность микросомии нижней челюсти коррелирует с общей тяжестью заболевания. Примерная частота встречаемости составляет 1:5600, причина неизвестна. Мужчины и женщины подвержены в равной степени. У трети пациентов аномалии развиваются на обеих половинах лица, у остальных 2/3 — только на одной. Правая и левая сторона поражаются одинаково часто. Окуло-аурико-вертебральная диспалазия является генетически гетерогенным синдромом. И хотя некоторые случаи синдрома Гольденхара были вызваны делециями в хромосомах 5р15 и 14q32, схожие фенотипы наблюдаются у пациентов с трисомиями 18,7,9 хромосом, а также терминальной делецией 22q. И хотя в большинстве случаев заболевание развивается спонтанно, генеалогические исследования показали, что возможен как аутосомно-рецессивный, так и аутосомно-доминантный тип наследования. С аналогичным фенотипом связаны и некоторые факторы со стороны матери: сахарный диабет, прием ретиноевой кислоты, талидомида, употребление кокаина. Поскольку поражаются обычно производные первые и второй жаберной дуги, к развитию классической клинической картины часто приводит разрыв стременной артерии во время внутриутробного развития. Схожесть проявлений окуло-аурикуло-вертебральной дисплазии и CHARGE-синдрома: (С [coloboma]—колобома глаз; Н [heart]—пороки сердца; A [atresia]—атрезия хоан, R [retarded growth] — задержка роста и развития; G [genital] — пороки развития мочеполовой системы, Е [ear] — глухота и пороки развития уха) позволяют предположить, что в основе патофизиологии синдрома лежит нарушение развития клеток нервного гребня. У детей с гемифациальной микросомией более чем в половине случаев имеются пороки других органов и систем (скелета, сердца, легких, центральной нервной системы, желудочно-кишечного тракта), причем их выраженность коррелирует с проявлениями микросомии. Нарушения формирования экстракраниального скелета имеются у 40-60% пациентов, наиболее часто поражаются позвоночный столб и ребра. У 25-50% детей с гемифациальной микросомией также имеются пороки сердца (наиболее часто —дефект межжелудочковой перегородки).
У 70-75% пациентов имеется кондуктивная или смешанная тугоухость. Возможно наличие микротии, атрезии наружного слухового прохода, мальформация или слияние слуховых косточек, отсутствие овального окна. Повышен риск воспалительных заболеваний среднего уха, часто эффективной оказывается установка тимпа-ностомических трубок на длительное время. В редких случаях встречается дисплазия улитки и полукружных каналов, ведущая к нейросенсорной тугоухости. Пациентам с окуло-аурикуло-вертебральным синдромом необходимо выполнять КТ височных костей перед любой операцией на среднем ухе, т.к. у них часто встречаются аномалии расположения лицевого нерва. Результаты хирургического лечения по поводу микротии или атрезии слухового прохода у детей с окуло-аурикуло-вертебральным синдромом обычно хуже, чем в изолированных случаях; это объясняется сочетанными нарушениями анатомии и роста костей лицевого скелета. Речь обычно развивается нормально, т.к. слух на одном ухе чаще всего сохранен. У пациентов с двусторонним снижением слуха используются традиционные слуховые аппараты или аппараты с проведением по кости. При выраженной двусторонней нейросенсорной тугоухости возможно проведение кохлеарной имплантации. Ключевым моментом лечения таких больных являются занятия с логопедом, т.к. слабость половины языка может вызывать нарушения артикуляции. Нарушения координации языка также могут вызывать проблемы с питанием. Все новорожденные, у которых во время кормления возникает кашель или поперхивания, должны обследоваться на предмет трахеопищевой фистулы, т.к. у данной группы пациентов отмечается высокая частота встречаемости. Краниофациальная реконструкция показана только в тяжелых случаях, обычно она ограничивается проведением дистракционного остеогенеза нижней челюсти. Также имеются сообщения о реконструкции верхней челюсти по Le Fort I и реконструкция нижней челюсти свободным лоскутом. Сведения о результатах зачастую разнятся. Во многих случаях для достижения приемлемого функционального и косметического результата требуется проведение нескольких оперативных вмешательств.
— Также рекомендуем «Причины врожденных аномалий наружного уха — ушной раковины, слухового прохода» Оглавление темы «Детская отоларингология»:
|
Источник