Синдром гарднера даймонда что это

Что такое синдром Гарднера?
Синдром Гарднера представляет собой редкий вариант семейного аденоматозного полипоза — состояние, характеризующееся множественными доброкачественными опухолями в толстой кишке и прямой кишке, которые в конечном итоге могут перерасти в колоректальный рак.
Термин «Гарднер» относится к ученому Элдону Дж. Гарднеру, который впервые описал синдром в 1951 году. Болезнь возникает из-за мутаций в гене Adenomatous Polyposis Coli (APC) — гене-супрессоре опухоли, который кодирует белки, ответственные за контроль деление и рост клеток. Мутация в этом гене запускает неконтролируемый рост клеток, что впоследствии приводит к образованию полипов или опухолей.
Синдром Гарднера может привести к изменениям в различных частях тела. Опухоли чаще всего появляются в толстой кишке, иногда в большом количестве. С возрастом они, как правило, увеличиваются. Помимо полипов в толстой кишке, синдром может вызывать появление фиброидов, десмоидных опухолей, сальных кист, сидячих полипов, гиперпластических полипов, кист нижней челюсти, брыжеечных кист. У некоторых пациентов с синдромом Гарднера могут встречаться даже поражения сетчатки глаза.
При синдроме Гарднера колоректальный рак (злокачественное новообразование толстого кишечника) развивается примерно в возрасте:
- 39 лет для людей, страдающих классическим семейным аденоматозным полипосом.
- 55 лет для людей, страдающих так называемым семейным аттенуированным аденоматозным полипосом.
Основным фактором риска развития синдрома Гарднера является наличие хотя бы одного родителя с этими заболеваниями. Спонтанная мутация в генах APC встречается гораздо реже.
Частота семейного аденоматозного полипоза, являющегося причиной синдрома Гарднера, колеблется от 1 на 7 000 до 1 на 22 000 человек.
Причины возникновения
Синдром Гарднера — это генетическое заболевание, а значит, наследственное. Ген Adenomatous Polyposis Coli (APC) является медиатором производства белков APC. Белок APC регулирует рост клеток, предотвращая их слишком быстрое или беспорядочное размножение.
У людей с синдромом Гарднера есть дефект гена APC. Это приводит к аномальному росту тканей. Что вызывает мутацию этого гена, пока не установлено.
Классификация синдрома Гарднера
Синдром Гарднера можно классифицировать следующим образом:
- классический тип семейного аденоматозного полипоза;
- семейный аттенуированный аденоматозный полипоз;
- семейный аденоматозный полипоз аутосомно-рецессивного типа.
У людей с классическим типом количество полипов увеличивается с возрастом до сотен тысяч, а семейный аттенуированный аденоматозный полипоз представляет собой, наоборот, вариацию развития расстройств, при которых происходит задержка роста полипа.
Люди с аутосомно-рецессивным типом заболевания страдают от более мягкой формы этого заболевания и имеют меньше полипов, чем люди с классическим типом. В последнем варианте развивается менее 100 полипов, что обусловлено мутациями в генах, отличительных от классического и семейного аттенуированного аденоматозного полипоза.
Особое значение имеют также нераковые образования, называемые десмоидными опухолями. Эти волокнистые опухоли обычно возникают в тканях, покрывающих кишечник, и могут быть спровоцированы операцией по удалению толстой кишки. Десмоидные опухоли имеют тенденцию повторяться и после хирургического удаления.
В каких ещё частях тела развивается синдром?
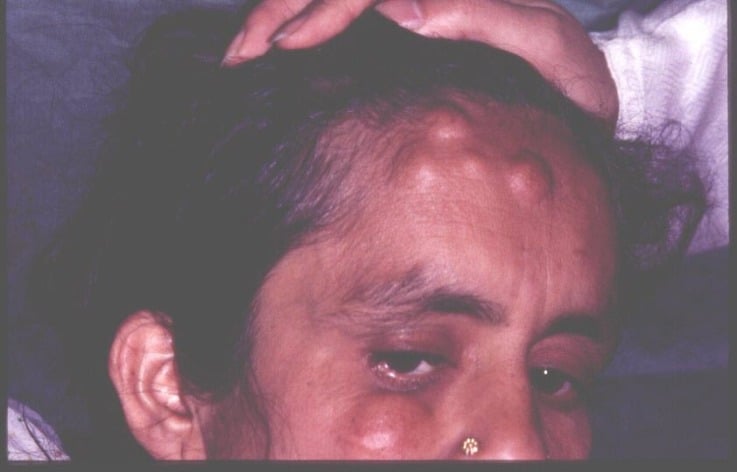
При синдроме Гарднера как при классическом, так и при аттенуированном полипозе доброкачественные и злокачественные опухоли могут развиваться в других частях тела, таких как:
- двенадцатиперстная кишка;
- желудок;
- кости;
- кожа.
Симптомы и признаки
Симптомы синдрома Гарднера у разных пациентов отличаются. Главной особенностью этого расстройства является наличие полипов толстой кишки, что проявляется у 80% — 99% пациентов.
Другие симптомы, связанные с синдромом Гарднера, включают в себя:
- полипы в желудке и тонкой кишке;
- зубные аномалии;
- доброкачественные новообразования костей или остеомы;
- доброкачественные опухоли кожи и соединительной ткани, называемые эпидермоидными кистами и десмоидными опухолями;
- пигментная эпителиальная гипертрофия сетчатки.
Диагностика
Несколько молекулярно-генетических тестов, которые обнаруживают мутацию в гене APC, доступны для диагностики синдрома Гарднера. Эти тесты включают генотипирование, исследование секвенирования, анализ числа копий генов и эпигенетическое тестирование.
Если семейный анамнез заболевания известен, анализ последовательности носителей является хорошим вариантом для будущих родителей, чтобы выяснить, являются ли они носителями вызывающей болезнь мутации. Это жизненно важный шаг на пути предотвращения передачи болезней следующему поколению.
Помимо генетического тестирования, проводиться рутинная диагностика синдрома Гарднера — это колоноскопия и рентгенологическое исследование длинных костей и челюстных костей. Обнаружение дополнительных признаков, таких как пигментные поражения в сетчатке, кожные аномалии и полипы в желудке и тонкой кишке, также подтверждает диагноз синдрома Гарднера.
Лечение синдрома Гарднера
Поскольку доброкачественные полипы в толстой кишке и прямой кишке появляются в среднем в возрасте 16 лет, регулярный скрининг пациентов начинается уже в возрасте 10 лет. Если симптомы не лечить, эти полипы могут превратиться в злокачественные опухоли в среднем в возрасте 40 лет.
Следовательно, ежегодный скрининг является лучшим способом предотвращения неблагоприятных последствий этого заболевания. Ежегодные осмотры с использованием колоноскопии, эзофагогастродуоденоскопии и обычных физических осмотров являются основной схемой лечения заболеваний.
Хирургическое удаление толстой кишки рекомендуется у пациентов с множественными запущенными полипами. Этим пациентам обычно назначают нестероидные противовоспалительные препараты (НПВП) для лечения оставшихся полипов.
Для десмоидных опухолей рекомендуется хирургическое удаление или лечение антиэстрогенами, НПВП, химиотерапия и радиотерапия. Эпидермоидные кисты обычно лечат так же, как обычные кисты, и иногда требуются удаления или инъекции стероидов.
Если присутствуют зубные аномалии, для устранения этих проблем может быть рекомендовано ортодонтическое лечение.
Как и при любых других заболеваниях, здоровый образ жизни с адекватным питанием, физическими упражнениями и снятием стресса может помочь людям справиться с физическими и эмоциональными проблемами.
Фармакотерапия
Такие препараты, как нестероидные противовоспалительные препараты (НПВП), могут помочь предотвратить прогрессирование колоректальных полипов до карциномы при семейном аденоматозном полипозе, одним из которых является синдром Гарднера.
Было показано, что они уменьшают размер и количество полипов в течение всего периода введения, но это не является неизменным, поскольку у некоторых пациентов одновременно появляются новые опухоли или злокачественная трансформация существующих полипов.
Более новые препараты в этой категории включают применение ингибиторов ЦОГ-2 поскольку рецепторы ЦОГ-2 присутствуют в слизистой оболочке толстой кишки, подвергаясь раковым изменениям. Результаты клинических испытаний показали, что размер полипов уменьшился, но влияние на уровень злокачественности не было определено.
Синдром Гарднера и рак
Кишечные полипы являются основной проблемой для пациентов с синдромом Гарднера из-за их склонности к раку. Однако полное удаление толстой кишки и большей части прямой кишки увеличивает 5-летнюю выживаемость пациентов почти до 100%.
Симптомы, такие как полипы кожи или остеомы, в первую очередь представляют косметическую проблему и должны лечиться, чтобы уменьшить видимый дискомфорт.
Какова перспектива жизни людей, затронутых этой болезнью?
Перспективы (прогноз) для людей с синдромом Гарднера варьируются, в зависимости от тяжести симптомов. Люди, у которых есть мутация гена APC, например, страдающие синдромом Гарднера, с возрастом все чаще заболевают раком толстой кишки.
Без хирургического лечения почти у всех людей с мутацией гена APC развивается рак толстой кишки к 39 годам (в среднем).
Источник
О. Л. Иванов, А. Н. Львов
«Справочник дерматовенеролога»
АУТОЭРИТРОЦИТАРНОЙ СЕНСИБИЛИЗАЦИИ СИНДРОМ (синдром Гарднера-Даймонда, психогенная пурпура) — васкулопатия аутоиммунного генеза с сенсибилизацией к компоненту стромы эритроцитов — фосфатидилесрину, характеризующаяся появлением на коже болезненных очагов инфильтрации, преобразующихся в течение суток в экхимозы.
Клиника.
Чрезвычайно редкое заболевание, встречается почти исключительно у женщин. Началу заболевания предшествуют небольшие механические травмы (ушибы), психоэмоциональные перегрузки, хирургические вмешательства, тяжёлая физическая работа. Первые ощущения пациента — чувство зуда и жжения на ограниченных участках кожного покрова, напоминающие «болезненный укус насекомого». Буквально через несколько минут появляется специфическое уплотнение кожи и подлежащих тканей. Очаги становятся видимыми через 4-5 часов и представляют собой болезненные отёчные эритематозные бляшки розово-красного цвета, размером от 3 до 10 см. Постепенно высыпания приобретают голубоватую (с желтовато-синюшным оттенком) окраску и в течение 1-1,5 суток принимают характер экхимозов. Эритема и отёк могут сохраняться около суток или более. При стихании воспалительно-инфильтративной реакции экхимозы становятся менее болезненными и, повторяя стадии «цветения синяка», подвергаются инволюции в течение 7-10 дней и не оставляют на коже никаких изменений.
Излюбленная локализация высыпаний — нижние конечности, особенно их вентральная поверхность, туловище. Иногда образование новых элементов сопровождается лихорадкой, болями в суставах, мышцах, головными болями, головокружением. Более чем у половины пациентов одновременно с кожными высыпаниями могут возникать боли в верхней части живота, желудочно-кишечные кровотечения, тошнота, рвота, поносы, гематурия, обильные менструальные кровотечения. Существенное место в общей картине заболевания занимают психоэмоциональные нарушения, с преобладанием в структуре личности истероидного радикала.
К достоверным признакам заболевания относится положительная диагностическая проба: при внутрикожном введении 1 мл 80% суспензии собственных отмытых эритроцитов пациента, через 24 часа на месте инъекции должны образоваться типичные болезненные воспалительно-инфильтративные очаги с последующим их переходом в экхимозы. Показатели коагулограммы — в пределах нормы.
Дифференциальный диагноз
Дифференциальный диагноз проводят с кожными проявлениями заболеваний, связанных с нарушениями внутрисосудистого свертывания (ДВС — синдром, тромбоцитопеническая геморрагическая пурпура), анафилактоидной пурпурой, узловатым ангиитом, спонтанным панникулитом, патомимией.
Лечение
Лечение не разработано.
Некоторый эффект оказывают адекватно подобранные психотропные средства, психотерапия.
Прогноз для жизни благоприятный.
Вернуться к списку статей о кожных заболеваниях
Статьи о некоторых других болезнях кожи:
Акродерматит атрофический хронический
О. Л. Иванов, А. Н. Львов
«Справочник дерматолога»Акродерматит пустулёзный стойкий Аллопо
О. Л. Иванов, А. Н. Львов
«Справочник дерматолога»Герпетиформный дерматоз Дюринга
Конспект лекции для студентов лечебного факультета.
Кафедра дерматовенерологии СПбГМАДерматоз субкорнеальный Снеддона-Уилкинсона
О. Л. Иванов, А. Н. Львов
«Справочник дерматолога»Лейшманиоз
О. Л. Иванов, А. Н. Львов
«Справочник дерматолога»Папилломатоз кожи карциноидный Готтрона
О. Л. Иванов, А. Н. Львов
«Справочник дерматолога»Папилломатоз сливной ретикулярный Гужеро-Карто
О. Л. Иванов, А. Н. Львов
«Справочник дерматолога»Полихондрит рецидивирующий
О. Л. Иванов, А. Н. Львов
«Справочник дерматолога»Фокса-Фордайса болезнь
О. Л. Иванов, А. Н. Львов
«Консультация дерматолога»Фолликулярный муциноз
Б. А. Беренбейн
«Дифференциальная диагностика кожных болезней»
Руководство для врачей.Фринодерма
О. Л. Иванов, А. Н. Львов
«Справочник дерматолога»
| Поиск по сайту «Ваш дерматолог» | |||
Источник

Синдром Гарднера – это наследственное заболевание, сопровождающееся полипозом толстого кишечника в сочетании с доброкачественными неоплазиями кожи, костей и мягких тканей. Может долгое время протекать бессимптомно. Возможны вздутие живота, урчание и расстройства стула. В некоторых случаях полипоз кишечника осложняется кровотечением или кишечной непроходимостью. Отмечается высокая вероятность развития колоректального рака. Заболевание диагностируется на основании жалоб, семейного анамнеза, данных осмотра, рентгенографии, КТ, МРТ, УЗИ, эндоскопии и других исследований. Лечение – эндоскопическая полипэктомия или резекция пораженных отделов кишечника.
Общие сведения
Синдром Гарднера – редкая генетически обусловленная патология, при которой наблюдается диффузный полипоз толстого кишечника в сочетании с доброкачественными опухолями костей и мягких тканей (остеомами, фибромами, нейрофибромами, эпителиальными кистами и другими неоплазиями). Полипозом при синдроме Гарднера преимущественно поражаются прямая и сигмовидная кишка, однако полипы могут выявляться в других отделах кишечника. Впервые был описан американским врачом и генетиком Е. Дж. Гарднером в 1951 году. С тех пор в специальной литературе появились упоминания более чем о ста случаях данного заболевания. Риск малигнизации полипов толстой кишки с развитием колоректального рака в течение жизни составляет около 95%. Лечение проводят специалисты в сфере клинической проктологии, гастроэнтерологии, онкологии, ортопедии, стоматологии и челюстно-лицевой хирургии.

Синдром Гарднера
Причины
Синдром Гарднера передается по аутосомно-доминантному типу. Выраженность кишечных и внекишечных клинических проявлений может сильно варьировать. Первые симптомы синдрома Гарднера обычно появляются у детей старше 10 лет. Возможно позднее начало с образованием первых опухолей в возрасте старше 20 лет. В отдельных случаях наряду с полипозом толстого кишечника, остеомами и мягкотканными новообразованиями у больных синдромом Гарднера обнаруживаются полипы тонкого кишечника, желудка и двенадцатиперстной кишки.
Симптомы
Синдром Гарднера включает в себя характерную триаду: диффузный полипоз нижних отделов толстого кишечника, остеомы плоских и трубчатых костей, различные доброкачественные опухоли кожи и мягких тканей. При умеренном количестве и небольшом размере полипов кишечные проявления синдрома Гарднера могут отсутствовать или быть слабо выраженными. В подростковом или юношеском возрасте больные обычно впервые обращаются к врачам в связи с появлением доброкачественных костных и мягкотканных опухолей.
Остеомы при синдроме Гарднера могут локализоваться как в плоских, так и в трубчатых костях. Часто наблюдается поражение костей лицевого черепа, сопровождающееся обезображиванием. Возможно смещение и даже выпадение зубов. Через некоторое время после появления рост остеом у больных синдромом Гарднера прекращается, опухоли не озлокачествляются. Неоплазии мягких тканей отличаются большим разнообразием. Особенно часто выявляются липомы, дерматофибромы, нейрофибромы и эпителиальные кисты. Реже встречаются атеромы, лейомиомы и другие новообразования. Мягкотканные опухоли при синдроме Гарднера также протекают доброкачественно, малигнизация отсутствует.
Полипы толстой кишки при синдроме Гарднера нередко становятся случайной находкой при проведении исследований ЖКТ по другим поводам либо обнаруживаются в процессе расширенного обследования, назначенного в связи с появлением множественных мягкотканных и костных неоплазий. В течении синдрома Гарднера можно выделить три стадии поражения кишечника. На первой стадии заболевание протекает бессимптомно. На второй пациенты отмечают дискомфорт в животе, вздутие, урчание и периодические нарушения стула. В каловых массах могут обнаруживаться примеси крови и слизи.
На третьей стадии у больных синдромом Гарднера выявляются выраженный болевой синдром, постоянный метеоризм, обильные примеси слизи и крови в испражнениях, снижение веса, повышенная утомляемость, эмоциональная лабильность, нарушения электролитного и белкового обмена. У многих пациентов с синдромом Гарднера развивается анемия, обусловленная небольшими по объему, но часто повторяющимися кровотечениями из нижних отделов ЖКТ. В отдельных случаях у больных развиваются неотложные состояния, требующие экстренной медицинской помощи – обильные кишечные кровотечения или кишечная непроходимость.
Диагностика
Диагноз устанавливается на основании семейного анамнеза (наличия синдрома Гарднера у близких родственников), клинической картины, включающей в себя характерную триаду, и данных дополнительных исследований. При проведении физикального осмотра врач отмечает наличие множественных костных и мягкотканных опухолей различной локализации. У некоторых больных синдромом Гарднера выявляются деформации лица, обусловленные остеомами лицевого черепа. При пальпации костей туловища и конечностей могут обнаруживаться опухолевидные образования костной плотности. При поражениях легкой степени количество неоплазий может быть незначительным, что затрудняет диагностику.
При пальпации живота наблюдается болезненность в левой подвздошной области. На первой стадии поражения кишечника данный симптом может отсутствовать. При проведении пальцевого ректального исследования на слизистой прямой кишки больных синдромом Гарднера обнаруживаются множественные узлы. На контрастных рентгеновских снимках такие узлы отображаются в виде дефектов наполнения. При узлах небольшого размера (менее 1 см) информативность контрастного рентгенологического исследования снижается. В ходе ректороманоскопии выявляются полипы в прямой и ободочной кишке. Количество полипов может сильно варьировать.
У некоторых пациентов с синдромом Гарднера отмечаются ограниченные поражения отдельных участков кишки. В отличие от рентгенографии, эндоскопическое исследование дает возможность диагностировать полипы любого размера, в том числе – мелкие (диаметром от 1-2 мм). Для уточнения характера и распространенности костных опухолей при синдроме Гарднера осуществляют рентгенографию. При мягкотканных новообразованиях назначают КТ, МРТ или УЗИ области поражения. При необходимости выполняют биопсию полипов, остеом и мягкотканных новообразований.
Дифференциальную диагностику синдрома Гарднера врачи-проктологи проводят с обычными множественными полипами и другими формами семейного полипоза. Для разных вариантов наследственного полипоза характерны определенные отличия в преимущественной локализации полипов (поражение всего толстого кишечника, поражение дистальных отделов толстой кишки), характере патологических изменений костей и мягких тканей. Для уточнения этих различий перед постановкой окончательного диагноза проводят детальный внешний осмотр, осуществляют ирригоскопию и колоноскопию.
Лечение синдрома Гарднера
Лечение только хирургическое. Поскольку риск озлокачествления костных и мягкотканных неоплазий отсутствует, решение о проведении оперативных вмешательств принимают при наличии косметического или функционального дефекта. Полипоз толстого кишечника при синдроме Гарднера рассматривается, как облигатный предрак, поэтому многие врачи считают целесообразным проведение операции до появления признаков малигнизации. При небольшом количестве полипов возможна эндоскопическая полипэктомия.
При синдроме Гарднера с выраженным диффузным полипозом показана резекция пораженного участка кишечника или тотальная колэктомия с наложением илеостомы либо формированием илеоректального анастомоза (при отсутствии полипов прямой кишки). Хирургическое вмешательство рекомендуют проводить в возрасте 20-25 лет. Из-за калечащего характера операции молодые пациенты с синдромом Гарднера нередко отказываются от данного вмешательства. В подобных случаях показано динамическое наблюдение с проведением колоноскопии через каждые 6-8 месяцев.
Некоторые врачи являются сторонниками выжидательной тактики и считают, что колэктомию при синдроме Гарднера следует проводить только при появлении признаков озлокачествления или при часто повторяющихся кровотечениях с развитием анемии. Показанием к экстренному оперативному вмешательству при синдроме Гарднера являются обильное кишечное кровотечение и кишечная непроходимость.
Прогноз и профилактика
При своевременном адекватном лечении прогноз при синдроме Гарднера достаточно благоприятный. Тяжесть течения определяется выраженностью полипоза и локализацией внекишечных опухолей. Родителям, имеющим родственников, страдающих данным заболеванием, в период планирования беременности рекомендуют обратиться за медико-генетической консультацией.
Источник