Синдром дефекта ногтей коленной чашечки
Синдром ногтей-надколенника
Синдром ногтей-надколенника (синдром nail-patella, NPS, остеониходисплазия, МКБ-10
— Q87.2) — редкое наследственное заболевание, которое характеризуется сочетанием
дисплазии ногтей, патологии костной системы и почек. Заболевание дебютирует в
младенчестве или детском возрасте. Лечение носит симптоматический характер. Прогноз
для пациентов определяется степенью поражения почек.
Синдром ногтей-надколенника связан с мутациями гена LMX1B.
Первое упоминание о больном с дистрофией ногтей и дисплазией скелета было сделано Э.
Шателайном (E. Chatelaine) в 1820 г. [1]. В 1930 г. У. Остеррайхер (W. Oesterreicher)
охарактеризовал триаду клинических симптомов синдрома ногтей-надколенника,
включающую в себя атрофию ногтей, аплазию надколенников и вывих головки лучевой
кости [2]. В 1950 г. К. Ф. Хокинс (C. F. Hawkins) и О. Е. Смит (O. E. Smith) установили,
что поражение почек является неотъемлемой частью данного синдрома [3].
Ген LMX1B, связанный с развитием синдрома ногтей-надколенника, был картирован в
1997 г. [4].
Распространенность и тип наследования
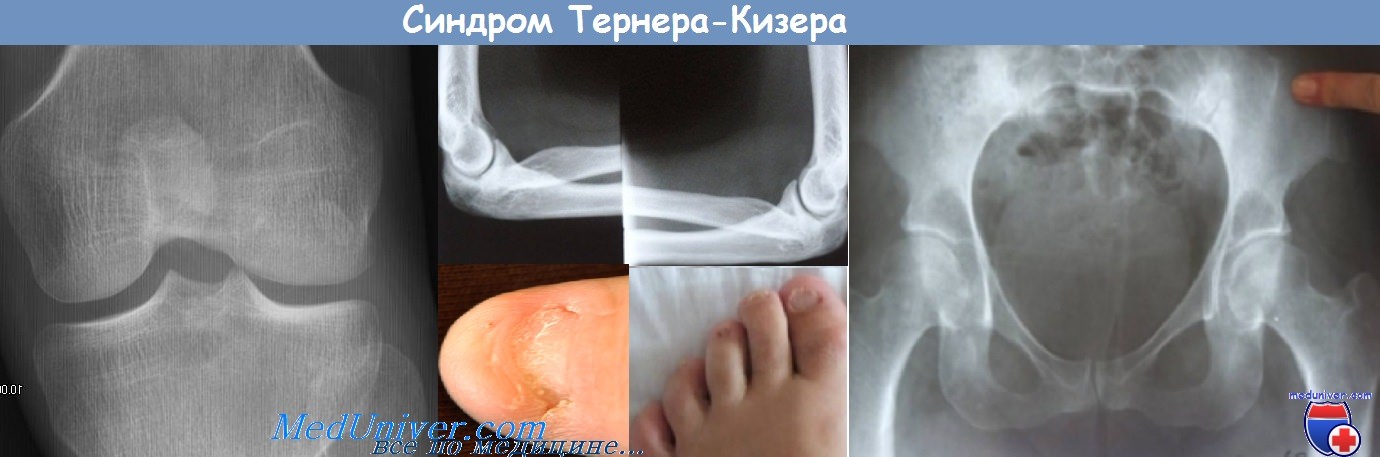
Заболевание дебютирует в младенчестве или детском возрасте. В 95 % случаев
врожденная патология ногтей при синдроме ногтей-надколенника проявляется в виде
гипоплазии или дисплазии ногтевых пластин (треугольные лунки, продольная
исчерченность, утолщение, губчатость) и в виде отсутствия ногтевой пластины. В 92 %
случаев выявляется гипоплазия и дисплазия надколенной чашечки, реже — ее отсутствие.
Нередко патология надколенника сопровождается недоразвитием латерального
надмыщелка бедренной кости, что ведет к рецидивирующим подвывихам надколенника. В
70 % случаев у пациентов имеются костные образования на внутренней поверхности
гребней подвздошных костей, так называемые «подвздошные рога». У многих больных
отмечается дисплазия костей предплечья в сочетании с гипоплазией головок плечевой и
лучевой костей, что приводит к повторным подвывихам локтевых суставов. У
большинства пациентов имеется дефицит массы тела независимо от характера питания,
наблюдаются поражения почек в виде аномалий развития органов мочевой системы,
протеинурии (присутствие белка в моче), изолированной или в сочетании с гематурией
(присутствие крови в моче). В 25 % случаев наблюдается нефропатия (поражение
клубочкового аппарата и паренхимы почек; у пациентов с данным синдромом —
аномалия гломерулярной базальной мембраны клубочков), в 10 % случаев развивается
хроническая почечная недостаточность. У пациентов встречаются варусная или
вальгусная деформации стоп, нарушения слуха, патологии органов зрения в виде
изменений роговицы (микрокорнеа — уменьшение размеров или уплощение роговицы,
склерокорнеа — частичная или полная «склеризация» роговицы (замещение ткани
роговицы склерой)), краевой пигментации радужки, врожденных катаракты и глаукомы
[7].
Первичную диагностику заболевания проводит педиатр. Диагноз ставится на основании
клинической картины, рентгенографии скелета, УЗИ почек, клинического анализа мочи,
консультации офтальмолога и отоларинголога. Диагноз подтверждается с помощью
молекулярно-генетического анализа LMX1B. Для данного заболевания возможна
пренатальная диагностика на основании биопсии ворсин хориона или амниоцентеза с
последующим молекулярно-генетическим анализом гена LMX1B [7].
В настоящее время специфическое лечение синдрома отсутствует, применяется
симптоматическая терапия. Для лечения и профилактики почечной недостаточности
применяются ингибиторы АПФ (ангиотензинпревращающего фермента). В терминальной
стадии хронической почечной недостаточности рекомендуется трансплантация почек [8].
Прогноз для пациентов определяется степенью поражения почек. Поражение ногтей при
синдроме ногтей-надколенника сохраняется на всю жизнь [8].
Синдром ногтей-надколенника связан с мутациями гена LMX1B [9]. Ген LMX1B кодирует
фактор транскрипции, играющий важную роль в развитии конечностей и почек. Белок
LMX1B регулирует экспрессию генов, кодирующих такие белки, как нефрин, подоцин,
CD2AP, а также цепи коллагена α3(IV) и α4(IV). Мутации гена приводят к нарушению
синтеза коллагена (основного структурного компонента гломерулярной базальной
мембраны почек) и дефекту подоцина, что связано с аномалиями ногтей и нарушением
функционирования почек [10].
- Ген LMX1B: c.176G>T (p.Cys59Phe)
- Ген LMX1B: c.305A>G (p.Tyr102Cys)
- Ген LMX1B c.306C>G (p.Tyr102Ter)
- Chatelaine (1820), quote by Roeckerath. W. Fortschritte auf der Gebiete der Rontgenstrahlen
1951: 75, 700–4. - Oesterreicher, W. Gemeinsame Vererbung von Anonychie bzw. Onychatrophie,
Patellardefekt und Luxatio radii. Dominantes Auftreten in 5 Generationen. Ztschr.
Konstitutionslehre 15: 465-476, 1930. - Hawkins, C. F., Smith, O. E. Renal dysplasia in a family with multiple hereditary
abnormalities including iliac horns. Lancet 255: 803-808, 1950. Note: Originally Volume I
[PubMed: 15416035] - Iannotti, C. A., Inoue, H., Bernal, E., Aoki, M., Liu, l., Donis-Keller, H., German, M. S.,
Permutt, M. A. Identification of a human LMX1 (LMX1.1)-related gene, LMX1.2: tissue-
specific expression and linkage mapping on chromosome 9. Genomics 46: 520-524, 1997
[PubMed: 9441763] - Sweeney, E., Fryer, A., Mountford, R., Green, A., McIntosh, I. Nail patella syndrome: a
review of the phenotype aided by developmental biology. J. Med. Genet. 40: 153-162, 2003
[PubMed: 12624132] - Renwick, J. H. Nail-patella syndrome: evidence for modification by alleles at the main locus.
Ann. Hum. Genet. 21: 159-169, 1956 [PubMed: 13373182] - Дружинина Т.В., Никонова О.П., Барсукова Т.И. Случай диагностики синдрома nail-
patella // Вестник Смоленской медицинской академии – 2010. — №4. – С. 78 – 81 - Константинова О.Д., Архипов В.В., Соловьев А.А., Майзельс И.Г.
Гормончувствительный вариант нефротического синдрома у ребенка с наследственной
остео-ониходисплазией // Нефрология – 2005. – Т.9. -№2. – С. 123 – 126 - Dreyer, S. D., Zhou, G., Baldini, A., Winterpacht, A., Zabel, B., Cole, W., Johnson, R. L.,
Lee, B. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail
patella syndrome. Nature Genet. 19: 47-50, 1998 [PubMed: 9590287] - Vollrath, D., Jaramillo-Babb, V. L., Clough, M. V., McIntosh, I., Scott, K. M., Lichter, P. R.,
Richards, J. E. Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-
patella syndrome. Hum. Molec. Genet. 7: 1091-1098, 1998. Erratum: Hum. Molec. Genet. 7:
1333 only, 1998 [PubMed: 9618165]
Источник
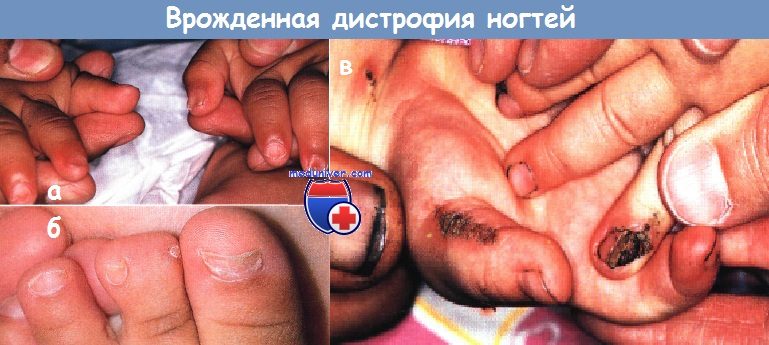
Врожденные и наследственные заболевания с нарушением роста ногтейОтсутствие, гипоплазия или дисплазия ногтей могут быть изолированными патологическими состояниями или же являться частью эктодермальной дисплазии или других генодерматозов. Аномалии ногтей: I. Анонихия: II. Наследственная эктодермальная дисплазия. III. Хромосомные аномалии: IV. Гиперпластические ногти: V. Койлонихия (ложкообразные ногти): VI. Медикаментозные мальформации: VII. Другие заболевания с дистрофией ногтей:
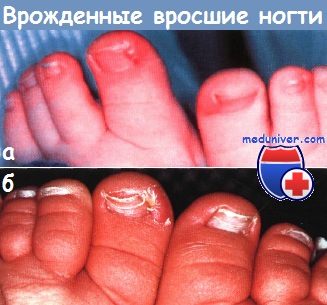
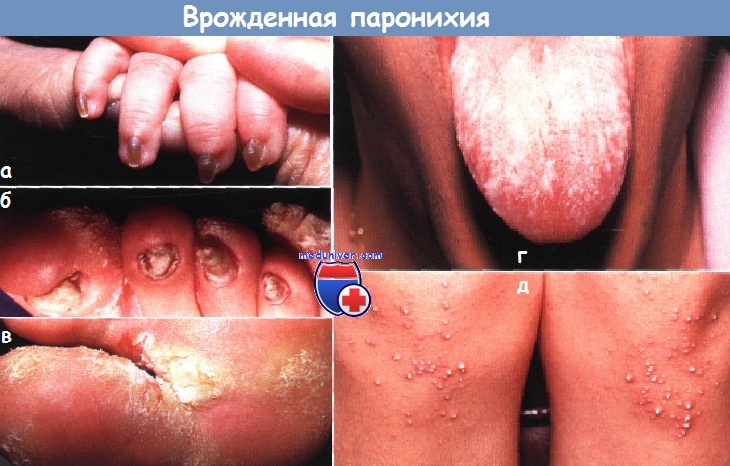
Поражения ногтей при изолированных мальформацияхАномальные ногти, изогнутые по типу часовых стекол (на пальцах типа «барабанные палочки»), и ложкообразные ногти могут быть наследуемыми по аутосомно-доминантному типу аномалиями без других сочетанных отклонений. Хотя врожденные вросшие ногти па пальцах стоп могут быть следствием врожденного смещения больших пальцев, которое корректируется только хирургическим путем, в большинстве случаев эти аномалии разрешаются самостоятельно и не связаны с анатомическим дефектом. Спонтанно регрессирующий вросший ноготь может быть результатом транзиторной гипоплазии ногтей стоп (особенно больших пальцев), которая обычно разрешается в пределах 12-18 мес. Изменения ногтей могут усиливаться вследствие внешней травмы и рецидивирующей/ хронической паронихии.
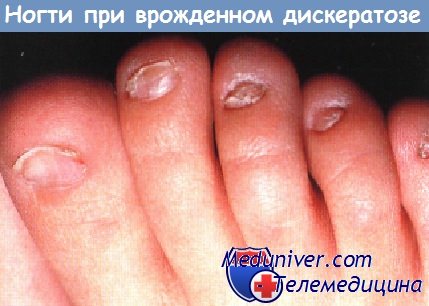
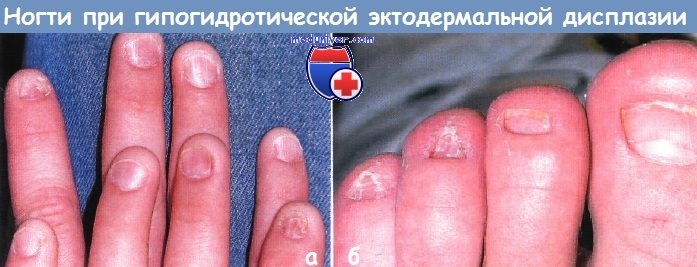
Поражения ногтей при эктодермальных дисплазияхМногие из эктодермальных дисплазии связаны с аномалиями ногтей; для некоторых из них они являются характерными и диагностически значимыми. При врожденной пахонихии, аутосомно-доминантной патологии с вариабельной пенетрацией, в первые несколько месяцев жизни развивается гиперкератоз ногтевого ложа. Затем происходит утолщение и удлинение ногтевой пластинки, приобретающей желтовато-коричневый цвет. Незначительная травма может привести к болезненному отделению ногтевой пластинки от ногтевого ложа и к кровотечению. Типичен гипергидроз ладоней и подошв, а также образование пузырей и мозолей. К другим признакам относятся лейкокератоз слизистой полости рта (не связанный со злокачественной дегенерацией), утолщение барабанных перепонок, приводящее к глухоте, леикокератоз роговицы и катаракты. Сообщалось также о гиперкератотических папулах на разгибательных поверхностях конечностей, дермоидных кистах и множественной стеатоцитоме. Врожденная пахонихия типа Ядассона-Левандовского, не связанная с глазными симптомами, вызывается мутацией в гене кератина 16 (KRT16) или в гене кератина 6А (KRT6A), расположенных в локусах 17q 12-21, 12q13. Дефекты в гене кератина 17 (KRT17) и кератина 6В (KRT6B) связаны с пахонихией по типу Джексона-Лоулера, при которой обнаруживается дистрофия сетчатки без лейкоплакии слизистой полости рта. Врожденный дискератоз можно ошибочно принять за врожденную пахонихию. Однако при врожденном дискератозе, который может быть сцепленным с Х-хромосомой, с аутосомно-доминантным или аутосомно-рецессивным типом наследования, ногтевая пластинка истонченная, наблюдаются продольные бороздки и птеригий. При врожденном дискератозе на шее и туловище выражена пойкилодермия с сетчатой пигментацией, телеангиэктазиями и атрофией. Лейкоплакия слизистой полости рта может ассоциироваться с развитием плоскоклеточного рака и другими злокачественными новообразованиями в ЖКТ. У 50% пациентов во второй и третьей декадах жизни отмечается панцитопения, которая напоминает анемию Фанкони. В большинстве случаев тип наследования рецессивный, сцепленный с Х-хромосомой, и ассоциируется с мутациями в локусе Xq28, в гене, кодирующем дискерин (DKC1). Гипоплазия ногтей может также быть признаком гидротической эктодермальной дисплазии и синдрома Коффина-Сириса.
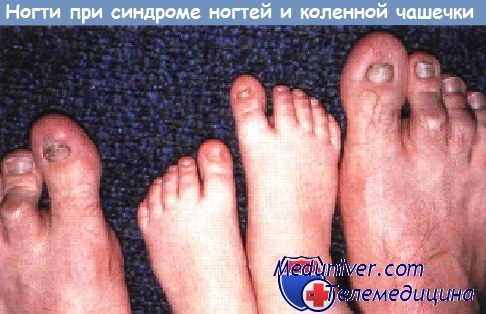
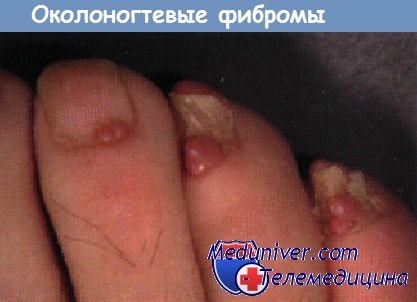
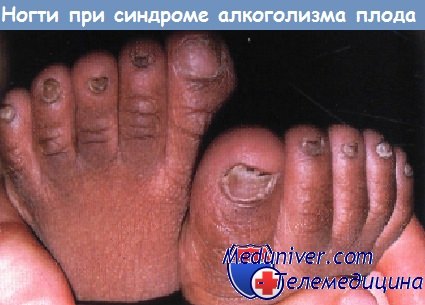
Поражения ногтей при генодерматозах и системных заболеванияхСиндром дефекта ногтей и коленной чашечки, аутосомно-доминантная аномалия, проявляется врожденным отсутствием или гипоплазией ногтей и коленной чашечки. В небольшом проценте случаев наблюдается гломерулонефрит, в редких случаях фатальный. Сообщалось также о гетерохромии радужной оболочки глаза, кератоконусе и катарактах. Околоногтевые фибромы, возникающие из проксимальной ногтевой бороздки, часто наблюдаются у пациентов с туберозным склерозом. Хотя фибромы появляются только в позднем детском или во взрослом возрасте, они могут стать первым ключом к диагнозу у лиц без других выраженных признаков поражения. Врожденная гипоплазия ногтей у детей, матери которых во время беременности принимали противосудорожные препараты, варфарин и другие тератогенные лекарственные средства, указывает на необходимость тщательной оценки ребенка на наличие других медикаментозно индуцированных признаков.
— Также рекомендуем «Паронихия у детей» Оглавление темы «Детская дерматология»:
|
Источник
Синдром ногтя-надколенника (остеоониходисплазия) — клиника, диагностикаДля этого аутосомно-доминантного заболевания характерны гипоплазия или отсутствие надколенника, ониходистрофия, дисплазия локтевых суставов, экзостозы крыла подвздошной кости и поражение почек. Установлено, что ген синдрома ногтя—надколенника находится на длинном плече 9-й хромосомы (сегмент 9q34). У мышей инактивация гена LMX1B, кодирующего фактор транскрипции гомеобокса LIM, приводила к аномалиям скелета (гипоплазия когтей и отсутствие надколенника) и дисплазии почек. Оказалось, что человеческий гомолог этого гена, мутации в котором были обнаружены при синдроме ногтя—надколенника, находится именно в сегменте 9q34. Считается, что ген LMX1B важен для нормального развития конечностей и почек, но точные механизмы влияния его мутаций на поражение почек остаются неясными. Клинические признаки поражения почек есть менее чем у половины больных. Прогноз благоприятный, лишь у 10% больных с поражением почек развивается терминальная почечная недостаточность. Клинические проявления: микрогематурия и легкая протеинурия, возникающие в подростковом или юношеском возрасте. У некоторых больных развивается нефротический синдром. Если возникает артериальная гипертония, то, как правило, легкая. При световой микроскопии и иммунофлюоресценции специфических изменений в почках нет, но их можно выявить при электронной микроскопии. В базальной мембране клубочка находят множественные просветления неправильной формы, как будто она изъедена молью. При специальном окрашивании в этих просветлениях обнаруживают фибриллы с характерной для коллагена исчерченностью. Эти фибриллы могут встречаться в базальной мембране клубочка у больныхс синдромом ногтя—надколенника даже в отсутствие клинических проявлений поражения почек, но в других базальных мембранах их нет. Данных о природе этих фибрилл нет. Исчерченные фибриллы, оказавшиеся коллагеном III типа, были обнаружены в базальной мембране клубочка у больных с изолированным поражением почек, без ониходистрофии или костных аномалий, в ряде случаев эта так называемая болезнь коллагена III типа носила семейный характер. Большинство заболевших — дети, у них быстро развивается терминальная почечная недостаточность. В отличие от синдрома ногтя—надколенника болезнь коллагена III типа наследуется аутосомно-рецессивно, что указывает на то, что это разные болезни. Специального лечения поражения почек при синдроме ногтя—надколенника нет. Трансплантация почки дает хорошие результаты, без рецидивов в трансплантате. Поскольку заболевание наследуется аутосомно-доминантно, необходимо проверять потенциальных доноров из числа родственников на внепочечные проявления болезни.
— Также рекомендуем «Кистозные болезни почек — типы, диагностика, лечение» Оглавление темы «Наследственные болезни почек»:
|
Источник
Н.С. ГЕРАСИМОВА,
учитель биологии муниципальной гимназии № 4,
г. Лыткарино, Московская обл.
Задачи по генетике человека
Задача 1
В медико-генетическую консультацию
обратился юноша (пробанд), страдающий глухотой. У
него есть сестра с нормальным слухом. Мать и отец
пробанда также имеют нормальный слух. У матери
пробанда пять сестер с нормальным слухом и один
брат, страдающий глухотой. Три сестры матери
пробанда замужем за здоровыми мужчинами. У одной
сестры матери пробанда растет здоровая дочь, у
второй – здоровый сын, у третьей – здоровая дочь
и глухой сын. Бабка пробанда но линии матери и ее
муж были здоровы. У бабки пробанда по линии
матери есть три здоровые сестры и два брата, один
здоровый, а другой глухой. Здоровые сестры бабки
по линии матери имели здоровых мужей, а здоровый
брат был женат на здоровой женщине. У первой
сестры бабки пробанда четыре здоровые дочери и
один глухой сын. У второй сестры бабки здоровая
дочь и глухой сын. У третьей сестры бабки
здоровая дочь и два сына, один здоровый, другой
глухой. Отец и мать бабки пробанда по линии
матери здоровы.
Заболевание наследуется по
аутосомно-рецессивному типу.
Определить, какова вероятность рождения
здоровых детей в семье пробанда, если он женится
на здоровой женщине, отец которой страдает тем же
недугом, что и пробанд. Составить
генеалогическое древо, определить вероятность
рождения здоровых детей.
Примечание. Глухонемота
связана с врожденной глухотой, которая
препятствует усвоению речи. Глухота может быть
звуковоспринимающего и звукопроводящего типов.
Наследственно обусловлено около половины всех
заболеваний глухонемотой, другая половинка –
фенокопии. Наследственные формы чаще передаются
рядом неаллельных аутосомных рецессивных генов.
Имеются аутосомно-доминантные и рецессивные
сцепленные с Х-хромосомой формы.
Решение
Ответ: половина детей будет
иметь нормальный слух, а половина – страдать
глухотой.
Задача 2
В медико-генетическую консультацию
обратилась молодая пара, собиравшаяся вступить в
брак, но обеспокоенная здоровьем будущих детей.
Их тревога объясняется тем, что молодые люди –
троюродные брат с сестрой. Юноша (пробанд)
страдает рахитом, который не излечивается
обычными дозами витамина D. Эта форма рахита
наследуется по доминантному типу, сцепленному с
полом. Сестра пробанда здорова. Мать – больна
рахитом, отец – здоров. У матери пробанда двое
братьев – оба здоровы. Дед пробанда по линии
матери болен, бабка здорова. Дед имел двух
братьев: здорового и больного. У здорового брата
деда от здоровой жены было два здоровых сына. У
больного брата деда жена была здорова, от их
брака родились две больные дочери и здоровый сын.
У одной больной дочери брата деда пробанда от
здорового мужа родилась здоровая дочь; у другой
больной дочери, состоящей в браке со здоровым
мужчиной, родились два сына, один из которых
болен, и больная дочь. У здорового сына брата деда
пробанда жена здорова, здоровы и их дети –
мальчики-близнецы.
Составить родословную, определить вероятность
рождения здоровых детей в семье пробанда, если он
вступит в брак со своей здоровой троюродной
сестрой.
Примечание. Рахит,
резистентный к витамину D (фосфат-диабет),
наследуется по доминантному типу, сцепленному с
полом. Клиническая картина сходна с рахитом.
Характерно искривление длинных трубчатых
костей, голеностопные и коленные суставы
деформированы. При отсутствии лечения дети
утрачивают способность ходить. Отмечается
необычно низкая концентрация неорганического
фосфора в крови.
Решение
Ответ: здоровыми могут родиться только
сыновья.
Задача 3
В медико-генетическую консультацию
обратился мужчина (пробанд), страдающий дефектом
ногтей и коленной чашечки. Его брат нормален.
Этот синдром имелся у отца пробанда, а мать была
здорова. Дедушка пробанда по линии отца имел
подобный синдром, а бабушка была здорова. Отец
пробанда имеет трех братьев и четырех сестер, из
них два брата и две сестры с синдромом дефекта
ногтей и коленной чашечки. Больной дядя по линии
отца женат на здоровой женщине и имеет здоровых
детей: двух дочерей и сына.
Составить генеалогическое древо; рассчитать
вероятность рождения в данной семье здорового
ребенка с IV группой крови с учетом того, что у
пробанда III группа крови, а его супруга имеет
II группу крови и не страдает дефектом ногтей и
коленной чашечки.
Примечание. У человека ген,
определяющий синдром дефекта ногтей и коленной
чашечки, – доминантный и находится в одной
хромосоме на расстоянии 10 морганид от гена
группы крови по системе АВО.
Решение
Ответ: в данной семье вероятность
рождения здорового ребенка с IV группой крови
составляет 2,5%.
Задача 4
В медико-генетическую консультацию
обратилась молодая семейная пара, обеспокоенная
здоровьем будущих детей. Их тревога объясняется
тем, что родители женщины были не вполне здоровы:
мать страдала ночной слепотой, отец – цветовой
слепотой.
Определить вероятность рождения здоровых детей.
Примечание. Гемералопия –
ночная, или куриная, слепота выражается в
отсутствии способности видеть при сумеречном
или ночном освещении; обычно – это составная
часть каких-либо синдромов. Чаще наследуется как
рецессивный сцепленный с Х-хромосомой признак.
Имеется аутосомно-рецессивный тип наследования.
Есть случаи аутосомно-доминантного наследования
ночной слепоты.
Частичная цветовая слепота называется
дальтонизмом (ахроматопией). Различают
протанопию – слепота на красный цвет,
дейтеронопию – слепота на зеленый цвет и
тританопию – слепота на синий цвет. Наследуется
как рецессивный сцепленный с полом признак. Есть
формы, наследуемые по аутосомно-рецессивному
типу.
Для Х-хромосомы человека с помощью рекомбинаций
локализовано 4 гена: цветовой слепоты (с),
гемофилии (h), мышечной дистрофии (m) и
куриной слепоты (n). Процент рекомбинаций
между с и h составляет 10%, между с и m –
25%, между с и n – 50%.
Решение
Ответ: в данной семье всегда
фенотипически здоровыми могут быть только
девочки.
Задача 5
В медико-генетическую консультацию
обратилась семейная пара, обеспокоенная
здоровьем будущих детей. Их тревога объясняется
тем, что оба супруга страдают легкой формой
талассемии. Кроме того, у женщины резус-фактор –
отрицательный, а у мужчины – положительный.
Определите вероятность рождения здорового
резус-отрицательного ребенка, если мать мужчины
была резус-отрицательной.
Примечание. Анемия
микроцитарная, или анемия Кули, или талассемия,
обусловлена расстройством синтеза нормального
«взрослого» гемоглобина. Кроме нарушения
морфологии эритроцитов (мишеневидная форма),
наблюдаются в различной степени выраженная
желтуха, изменения в скелете и др. Гомозиготы в
90–95% случаев гибнут в раннем возрасте, у
гетерозигот талассемия протекает в относительно
легкой форме. Наследование аутосомное с неполным
доминированием.
Резус-фактор – один из множества антигенных
свойств крови. В простейшем варианте
резус-положительность доминирует над
резус-отрицательностью.
Решение
Ответ: вероятность рождения
здорового резус-отрицательного ребенка в данной
семье равна 1/8.
Источник