Синдром де груши этиология признаки методы диагностики лечение прогноз

Причина.
Делеция короткого плеча 18-й хромосомы
(возможны транслокационные и мозаические
варианты).
Клиника.
Низкая масса тела при рождении, профиль
плоский или вогнутый. Маленький рост,
микроцефалитная форма черепа, круглое
лицо, высокое небо (иногда с расщелиной).
Очаги облысения на голове либо тотальная
алопеция. Характерна деформация зубов
и ушных раковин, пупочечные и паховые
грыжи. Аномалия кистей рук и пальцев,
синдактилия пальцев ног. У мальчиков
часто недоразвитие половых органов.
Отмечается
умственная отсталость.
Патогенез.
У больных с грубой мозговой патологией
резкое снижение продолжительности
жизни. Косоглазие, мышечная гипотония,
гипоплазия полового члена и мошонки у
мальчиков и гипоплазия малых половых
губ у девочек. Пороки сердца, иногда
почек.
Диагностика.
Исследование кариотипа, цитологическое
обследование.
Лечение.
Симптоматическое.
Частота.
1: 60000.
Синдром Лежена
Причина.
Делеция длинного плеча 18-й хромосомы.
Клиника.
Синдром мышечной гипотонии. Ребенок
лежит на спине в позе «лягушки». Череп
микроцефальной формы, уплощенное лицо
с выступающим подбородком. Косоглазие,
птоз, нистагм, снижение зрения, высокое
твердое небо (иногда с расщелиной),
своеобразная форма ушных раковин,
нередко сужение слуховых органов.
Интеллектуальное
нарушение варьируется от легкой до
олигофрении в степени идиотии.
Патогенез.
Пороки сердца, иногда почек. Пороки
развития зрительной системы.
Диагностика.
Дерматоглифика (повышенное число
завитков на пальцах рук), цитологическое
обследование.
Частота.
1:60000.
Синдром Реторе
Причина.
Частичная трисомия по короткому плечу
хромосомы 9.
Клиника.
Череп у новорожденных микробранхицефальный
с уплощенным затылком. С возрастом
брахицефалия уменьшается. Роднички
широко открыты, имеется лобный шов.
Характерны глазные аномалии: микро- или
энофтальмия, страбизм, нарушение
рефракции, эпикант, антимонголоидный
разрез глаз, крупный нос с широким
кончиком, опущены углы рта, короткая
верхняя губа, «конские» зубы, дипластичное
телосложение. Умственная
отсталость варьируется от легкой до
глубокой.
Патогенез.
Глазные аномалии, у 25%- врожденные пороки
сердца. Двигательные расстройства,
нарушение координации.
Диагностика.
Дерматоглифика, цитологическое
обследование, кариологическое
исследование.
Лечение.
Отсутствует.
Синдром Брадера – Вилли
Причина.
Утраивается участок 15-й хромосомы
отцовского происхождения
Клиника.
Мышечная гипотония, половое недоразвитие,
ожирение, умственная отсталость. Низкая
масса тела при рождении, пониженная
температура. Потом развивается
чрезвычайный аппетит. Деформированные
низко расположенные ушные раковина и
мягкие ушные хрящи, подковообразная
форма рта, короткая губа, неправильный
рост зубов. Диспропорциональность стоп
и кистей. Нарушение осанки.
Патогенез.
Частые грыжи, патология кистей и стоп.
В пубертатном возрасте диабет. Взрослые
страдают гиперсомнией, ишемической
болезнью сердца, инфаркт миокарда.
Диагностика.
Клиническое обследование.
Лечение.
Симптоматическое.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник
Синдром «кошачьего крика»
Причина.
Делеция короткого плеча 18-й хромосомы
(возможны транслокационные и мозаические
варианты).
Клиника.
Низкая масса тела при рождении, профиль
плоский или вогнутый. Маленький рост,
микроцефалитная форма черепа, круглое
лицо, высокое небо (иногда с расщелиной).
Очаги облысения на голове либо тотальная
алопеция. Характерна деформация зубов
и ушных раковин, пупочечные и паховые
грыжи. Аномалия кистей рук и пальцев,
синдактилия пальцев ног. У мальчиков
часто недоразвитие половых органов.
Отмечается
умственная отсталость.
Патогенез.
У больных с грубой мозговой патологией
резкое снижение продолжительности
жизни. Косоглазие, мышечная гипотония,
гипоплазия полового члена и мошонки у
мальчиков и гипоплазия малых половых
губ у девочек. Пороки сердца, иногда
почек.
Диагностика.
Исследование кариотипа, цитологическое
обследование.
Лечение.
Симптоматическое.
Частота.
1: 60000.
Синдром Лежена
Причина.
Делеция длинного плеча 18-й хромосомы.
Клиника.
Синдром мышечной гипотонии. Ребенок
лежит на спине в позе «лягушки». Череп
микроцефальной формы, уплощенное лицо
с выступающим подбородком. Косоглазие,
птоз, нистагм, снижение зрения, высокое
твердое небо (иногда с расщелиной),
своеобразная форма ушных раковин,
нередко сужение слуховых органов.
Интеллектуальное
нарушение варьируется от легкой до
олигофрении в степени идиотии.
Патогенез.
Пороки сердца, иногда почек. Пороки
развития зрительной системы.
Диагностика.
Дерматоглифика (повышенное число
завитков на пальцах рук), цитологическое
обследование.
Частота.
1:60000.
Синдром Реторе
Причина.
Частичная трисомия по короткому плечу
хромосомы 9.
Клиника.
Череп у новорожденных микробранхицефальный
с уплощенным затылком. С возрастом
брахицефалия уменьшается. Роднички
широко открыты, имеется лобный шов.
Характерны глазные аномалии: микро- или
энофтальмия, страбизм, нарушение
рефракции, эпикант, антимонголоидный
разрез глаз, крупный нос с широким
кончиком, опущены углы рта, короткая
верхняя губа, «конские» зубы, дипластичное
телосложение. Умственная
отсталость варьируется от легкой до
глубокой.
Патогенез.
Глазные аномалии, у 25%- врожденные пороки
сердца. Двигательные расстройства,
нарушение координации.
Диагностика.
Дерматоглифика, цитологическое
обследование, кариологическое
исследование.
Лечение.
Отсутствует.
https://www.youtube.com/watch?v=YtKn9onXpGQ
Причина.
Утраивается участок 15-й хромосомы
отцовского происхождения
Клиника.
Мышечная гипотония, половое недоразвитие,
ожирение, умственная отсталость. Низкая
масса тела при рождении, пониженная
температура. Потом развивается
чрезвычайный аппетит. Деформированные
низко расположенные ушные раковина и
мягкие ушные хрящи, подковообразная
форма рта, короткая губа, неправильный
рост зубов. Диспропорциональность стоп
и кистей. Нарушение осанки.
Патогенез.
Частые грыжи, патология кистей и стоп.
В пубертатном возрасте диабет. Взрослые
страдают гиперсомнией, ишемической
болезнью сердца, инфаркт миокарда.
Диагностика.
Клиническое обследование.
Синдром Ретта – генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника.
Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного.
Общие сведения
Синдром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году.
Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии.
Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера.
Синдром Ретта
Этиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается.
Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани.
При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей.
Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК.
Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко.
Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца.
У новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям.
Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе.
Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения.
Третья стадия синдрома Ретта называется псевдостационарной, так как при ней мало заметны признаки прогрессирования заболевания. Обычно она длится от 4 до 15 лет, состояние больных стабильно, однако наблюдаются судорожные приступы, глубокая умственная отсталость, гиперкинезы. В большинстве случаев синдрома Ретта этот этап оканчивается в пубертатном периоде.
Четвертая стадия синдрома Ретта характеризуется уменьшением частоты эпилептических припадков вплоть до их исчезновения при прогрессировании двигательных расстройств. Большинство больных полностью теряют подвижность, возникает атрофия мышц, сосудистые нарушения в нижних конечностях, что может привести к развитию трофических язв. Из-за слабости мышечного корсета спины при синдроме Ретта возникает сколиоз или другие формы искривления позвоночника.
Поиск Лекций
Источник
В качестве примера синдрома со структурной перестройкой хромосом можно привести синдром делеции короткого плеча 5-й хромосомы — синдром 5р-, или синдром «кошачьего крика». Среди больных этим синдромом преобладают девочки. Частота встречаемости — 1:50 000 новорожденных. Характерные симптомы — это микроцефалия, круглое лицо (с возрастом оно вытягивается), широко расставленные глаза, антимонголоидный разрез глаз, эпикант, недоразвитие нижней челюсти, катаракта, косоглазие и другие глазные нарушения.
Синдром получил свое название в связи с тем, что крик новорожденных с этим синдромом напоминает кошачье мяуканье. Выраженная умственная отсталость отмечается во всех случаях. Среди детей с глубокой умственной отсталостью на эту патологию приходится 1% из всех случаев синдрома.
Трисомия-18
К синдромам с анеуплоидией относится трисомия-18, или синдром Эдвардса, встречающийся с частотой 1:7000. В данном случае лишней является хромосома 18-й пары. При этом синдроме преобладают девочки. Характерные симптомы синдрома — выступающий затылок, тонкие переносье и спинки 1, недоразвитие нижней челюсти, «птичий профиль», деформированные ушные раковины и тяжелые пороки развития внутренних органов (чаще всего сердца), в связи с чем такие дети умирают в раннем возрасте. До года доживают 10% детей с трсомией-18. Все больные отстают в умственном и физическом развитии.
Известны (описаны) и другие аномалии хромосомы-18, включающие как структурные, так и геномные перестройки.
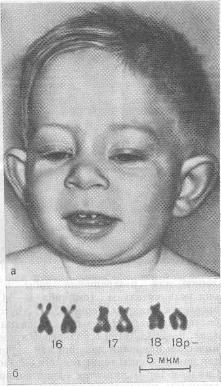
Синдром де Груши
Моносомия-18р. Частота встречаемости — 1: 60 000. Обычно ребенок рождается в срок, но с небольшой массой тела. В дальнейшем характерными признаками являются: маленький рост, микроцефальной формы череп, круглое лицо. Часто имеют место очаги облысения на голове либо тотальная алопеция. Характерны деформация зубов и ушных раковин, не редкость пупочных и паховых грыж на фоне мышечной гипотонии, аномалии кистей руки пальцев, синдактилия пальцев ног, «стопа-качалка». У мальчиков часто бывает недоразвитие половых органов, характерно резкое снижение продолжительности жизни у больных с грубой мозговой патологией. Реже наблюдаются более легкие формы интеллектуального дефекта с нормальной продолжительностью жизни. Характерно сочетание умственной отсталости с судорожным синдромом и различными речевыми расстройствами. Помимо делеции, возможны транслокационные и мозаичные варианты.
Синдром Лежена
Моносомия-18q, или синдром 18q — синдром Лежена. Делеция длинного плеча хромосомы-18. Встречается с частотой 1: 60 000. Девочки с этим синдромом рождаются в 1,5 раза чаще, чем мальчики. Одним из ранних характерных признаков считается синдром мышечной гипотонии: ребенок лежит на спине в «позе лягушки». Обращают на себя внимание микроцефальной формы череп, уплощенное лицо с выступающим подбородком. Характерна частота различных пороков развития зрительной системы, т.е. при данном синдроме имеет место дефект развития, включающий дефекты зрения и интеллекта. Среди нарушений зрительной функции преобладают колобомы, косоглазие, птоз, нистагм, снижение остроты зрения, атрофия зрительных нервов. Характерны своеобразная форма носа, рта, высокое твердое нёбо, иногда с расщелиной, своеобразная форма ушных раковин, нередко сужение или атрезия наружных слуховых проходов. Характерным признаком считается недоразвитие наружных половых органов, нередко отмечаются пороки сердца, почек. Интеллектуальные нарушения варьируются от легкой ^пограничной интеллектуальной недостаточности и даже нормального интеллекта) до олигофрении в степени идиотии.
Синдром Патау
К геномным мутациям относится также трисомия-13 — синдром Патау. Встречается синдром с частотой 1:6000 новорожденных. Дети рождаются с истинной пренатальной гипотрофией. В 50% случаев беременность осложняется многоводием. Типичным признаком является расщелина губы и неба. Как правило, дети страдают полидактилией. Характерны пороки развития органов зрения: катаракта, микрофтальмия, анофтальмия, циклопия. Череп неправильной формы, возможна тригоноцефалия, узкие глазные щели, запавшее переносье, деформированные низко расположенные ушные раковины, дефекты скальпа. Отмечаются рефлексорное сгибание костей, «стопа-качалка», пороки внутренних органов: врожденные пороки сердца, почек, желудочно-кишечного тракта, мочеполовой системы.
Продолжительность жизни резко снижена <Ф5% детей погибает в возрасте до года). В развитых странах отмечается тенденция к увеличению продолжительности жизни таких больных: 15% детей доживают до пятилетнего возраста и 2—3% — до десятилетнего возраста. Во всех случаях отмечается выраженное психическое недоразвитие.
Помимо трисомной (75%), встречаются транслокационная (20%) и мозаичная (5%) формы.
Синдром Реторе
С различными типами хромосомных аномалий может быть связана трисомия-9р — синдром Реторе. Предполагается, что в группе умственно отсталых детей трисомия по короткому плечу 9-й хромосомы занимает по частоте второе место после
рома Дауна. Девочки с данным синдромом встречаются в два раза чаще, чем мальчики. Череп у новорожденных микробранхицефальный с уплощенным затылком. С возрастом брахицефалия уменьшается. Роднички широко открыты, имеется лобный шов. Характерны глазные аномалии: микро- или энофтальмия, страбизм, нарушения рефракции, гипертелоризм эпикант, антимонголоидный разрез глаз, крупный нос с широким кончиком, опущенные углы рта, короткая верхняя губа, «конские» зубы, диспластичное телосложение. У 25% детей врожденные пороки сердца. Умственная отсталость диагносцируется у всех больных. Выраженность интеллектуального дефекта варьируется от легкой до глубокой; типичны эмоциональная лабильность, повышенная психомоторная возбудимость, двигательные расстройства с нарушением координации движений. Среди цитогенетических вариантов синдрома Реторе — транслокационный, связанный с частичной трисомией (иногда с тетрасомией), «свободная» трисомия, мозаицизм предполагается, что характерные фенотипические проявления связаны с сегментом 9р21.
Читайте также:
Рекомендуемые страницы:
©2015-2020 poisk-ru.ru
Все права принадлежать их авторам. Данный сайт не претендует на авторства, а предоставляет бесплатное использование.
Дата создания страницы: 2016-02-12
Нарушение авторских прав и Нарушение персональных данных
Поиск по сайту:
Источник