Синдром беквита видемана по мкб

Содержание
- Описание
- Причины
- Симптомы
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Синдром Видемана-Беквита.
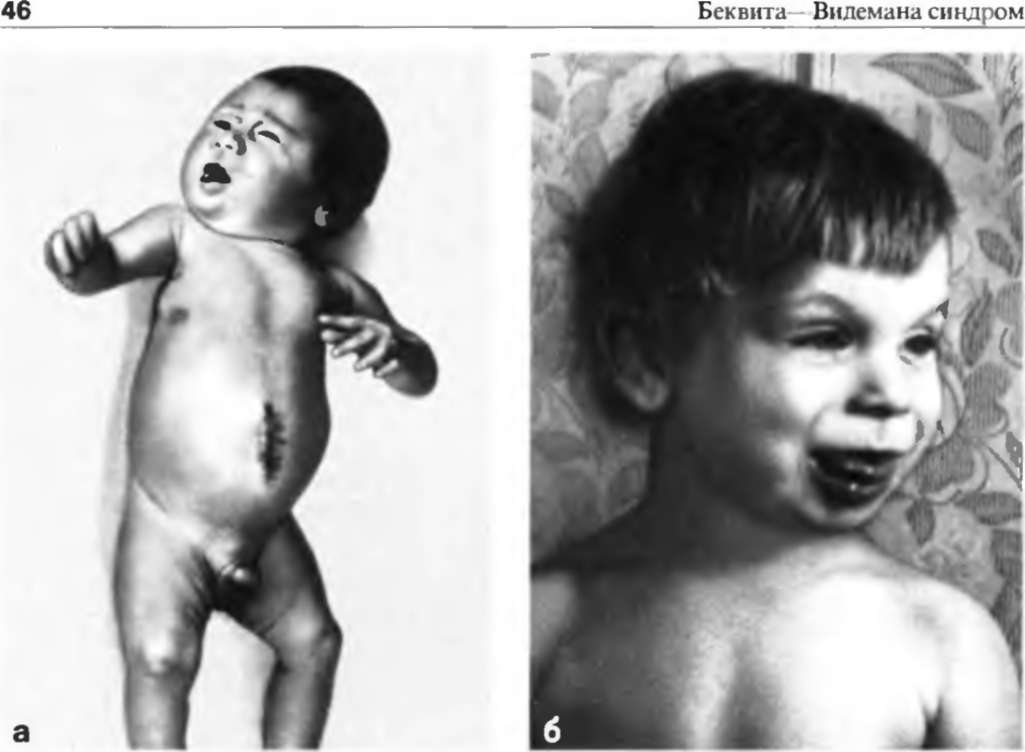
Ребенок с синдромом Видемана-Беквита
Описание
Синдром Беквита-Видемана (Beckwith-Wiedemann) впервые был описан J. В. Beckwith в 1963 году и H. R. Wiedemann в 1964 году. Этот синдром характеризуется классической триадой, включающей макросомию, омфалоцеле и макроглоссию.
Синонимы. Синдром экзомфалии — макроглоссии — гигантизма.
Частота встречаемости оценивается на уровне 0,72 на 10 000 родов. В литературе описано более 500 клинических наблюдений этого заболевания.
Причины
В большинстве случаев синдром Беквита-Видемана (Beckwith-Wiedemann) возникает спорадически и имеет аутосомно-рецессивный тип наследования с неполной пенетрантностью и вариабельной экспрессивностью. Предполагается, что данное заболевание может возникать вследствие перестроек вовлекающих регион короткого плеча хромосомы 11 р15.
Симптомы
Название «EMG-синдром» происходит из трех наиболее манифестных его признаков – экзомфалос (E), макроглоссия (M) и гигантизм (G). Среди свойственных синдрому признаков основными являются макроглоссия, пуповинная грыжа и другие пупочные аномалии, а также гипогликемия.
Макроглоссия выявляется в любом возрасте ребенка, часто отмечается с рождения. Язык может не помещаться во рту, за счет чего рот ребенка открыт, а лицо напоминает таковое у больного с гипотиреозом. Увеличенный язык затрудняет сосание и даже дыхание новорожденного, у более старших детей отмечают дизартрические расстройства.
Гипогликемия у новорожденного манифестирует уже на первые-третьи сутки. Развивающиеся за счет этого коматозные состояния могут повлечь за собой смерть ребенка на первом году жизни или тяжелое органическое поражение мозга, сопровождающееся умственной отсталостью. Развитие гипогликемических состояний связано с гиперплазией островковых клеток поджелудочной железы, приводящей к гиперинсулинемии. С начинающейся еще внутриутробно гиперпродукцией инсулина, обладающего анаболическим действием, связывают как макроглоссию, макросомию, висцеромегалию, так и предрасположенность к развитию опухолей паренхиматозных органов. Явления гипергликемии самопроизвольно убывают в течение первых месяцев жизни больного.
Склонность к увеличению массы тела отмечается уже при рождении, она обычно превышает 4000 г, а длина – 52 Внутриутробная висцеромегалия, по-видимому, является причиной образования различных грыж, в том числе характерного для синдрома омфалоцеле (пуповинной грыжи). Пуповинная грыжа диагностируется у новорожденного и может быть различной по величине, иногда достигая размеров детской головки.
Макросомия с увеличением мышечной ткани и подкожного жирового слоя отмечается с рождения или развивается постнатально. Постнатальный гигантизм относится к менее постоянным признакам, иногда проявляется увеличением одной половины тела.
Лечение
Медикаментозное лечение: специфическое лечение гиперинсулинизма у новорождённых.
Хирургическое лечение эмбриональных неоплазий по стандартным методикам.
Прогноз
Синдром Беквита-Видеманна имеет разный прогноз для жизни. Он определяется своевременной диагностикой гипогликемии (профилактика умственной отсталости) и ранней диагностикой эмбриональных опухолей.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Синдром Беквита — Видемана (англ. BWS — Beckwith-Wiedemann-Syndrom, EMG — Exomphalos-Makroglossie-Gigantismus, также синдром «гигантизма с пуповинной грыжей») — одна из редких генетических аномалий, связана с превышением норм роста плода при беременности и с несогласованным развитием различных отделов организма.
Типичными признаками синдрома являются увеличенные размер и вес плода (новорождённого ребёнка), причем происходят не только асимметричный рост организма и внешние отклонения в размерах тела, но и непропорционально большой размер некоторых внутренних органов: печени, селезёнки, почек и языка.[1]
Особенности синдрома[править | править код]
На наличие синдрома может указывать наличие одного или более из следующих факторов:
- Пренатальное и постнатальное опережение роста (таковым обычно считают рос и вес выше 97 процентили)
- Макроглоссия
- Омфалоцеле (грыжа пупочного канатика, пуповинная грыжа, эмбриональная грыжа — грыжевой мешок, сформированный брюшиной, выходит из брюшной полости через пупочное отверстие (пупок))
- Висцеромегалия
- Эмбриональные опухоли (такие как гепатобластома, нефробластома, рабдомиосаркома) в детском возрасте
- Гемигиперплазия (асимметричный рост одной или нескольких частей тела)
- Почечные аномалии
- Адренокортикальная цитомегалия
- Неонатальная гипогликемия
Частые симптомы[править | править код]
- Лёгкая или умеренная умственная отсталость (с неуточнённой частотой).
- Рост:
- Макросомия с увеличением мышечной массы и подкожного жирового слоя;
- ускоренное созревание костной ткани;
- широкие метафизы и узкие диафизы длинных трубчатых костей;
- слабо заметный переход проксимального метафиза плечевой кости в диафиз.
- Голова:
- макроглоссия;
- экзофтальм — смещение глазного яблока вперёд (выпученные глаза) с относительным недоразвитием подглазничного края;
- пламенеющий невус на лбу и веках;
- выступающий лобный шов;
- широкие роднички;
- выступающий затылок;
- неправильный прикус из-за нижней прогнатии (выступающая вперед нижняя челюсть) и верхней микрогнатии (врождённое недоразвитие челюстной кости);
- насечки на мочках ушей и задней поверхности завитков.
- Внутренние органы:
- нефромегалия (увеличение размеров одной или обеих почек) с дисплазией (неправильным развитием) мозгового вещества почек;
- панкретомегалия (увеличение размеров поджелудочной железы) с гиперплазией (размножением клеток и образованием новых тканевых структур) островков поджелудочной железы;
- гипертрофия (увеличение объёма и массы) клеток коры надпочечников у плода (постоянный признак);
- гиперплазия интерстициальных клеток половых желез (межуточных клеток, расположенные в строме яичников и между канальцами семенников у млекопитающих; участвуют в выработке половых гормонов: в семенниках — андрогенов, в яичниках — эстрогенов[2]) и амфифильных клеток аденогипофиза.
- Прочее:
- эритроцитоз (увеличение количества эритроцитов) у новорождённых;
- гипогликемия в раннем детском возрасте (30-50 %);
- грыжа пупочного канатика и другие аномалии пуповины; расхождение прямых мышц живота;
- релаксация диафрагмы;
- крипторхизм (неопущение яичка в мошонку);
- врождённые пороки сердца, в том числе изолированная кардиомегалия.[3]
Этиология и встречаемость болезни[править | править код]
Синдром Беквитта-Видемана — панэтнический (не связанный с принадлежностью к какой-либо национальности) синдром. Возникает спорадически, иногда наследуется по аутосомно-доминантному типу. Встречается приблизительно у одного из 13 700 родившихся живыми детей[1].
Синдром Беквитта-Видемана вызван нарушением баланса экспрессии импринтированных генов в регионе р15 хромосомы 11[1].
Среди генов, расположенных к этом регионе, находятся кодирующие белки гены CDKN1C и IGF2:
- CDKN1C кодирует супрессор клеточного цикла, ограничивающий деление и рост клеток;
- IGF2 кодирует инсулиноподобный фактор роста, стимулирующий рост.
Также в этом регион расположены транскрибируемые, но не транслируемые KCNQOT1 и Н19. Их транскрипция подавляет экспрессию отцовской копии CDKN1С и материнской копии IGF2 соответственно: в норме эти гены импринтированы и экспрессируются только из отцовского (IGF2 и KCNQOT1) или только материнского аллеля (HI9 и CDKN1C)[1].
Ссылки[править | править код]
- Безуглаяя А. А. Синдром Беквита — Видемана
- О синдроме на сайте medaboutme.ru
Примечания[править | править код]
Источник
Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Беквит-Видемана синдром (BWS) врожденное заболевание, которое характеризуется чрезмерно быстрым ростом в младшем возрасте, ассиметрией развития тела, повышенным риском развития рака и некоторых врожденных дефектов, нарушением поведения ребенка. Впервые описано как семейная форма омфалоцеле с макроглоссией в 1964 году, немецким доктором Ханс-Рудольф Видеманом. В 1969 году, Дж. Брюс Беквит из Университета Лома Линда, штат Калифорния, описал подобные симптомы у нескольких пациентов. Первоначально, профессор Видеман ввел термин синдром EMG, чтобы описать сочетание врожденной пупочной грыжии, макроглоссии и гигантизма. Со временем, эта патология была переименована в синдром Беквит-Видемана (BWS).
[1], [2], [3], [4]
Код по МКБ-10
Q87.3 Синдромы врожденных аномалий, проявляющиеся избыточным ростом [гигантизмом] на ранних этапах развития
Эпидемиология
Синдром Беквита-Видеманна встречается с частотой 1 на 13 700 новорождённых.
[5], [6], [7], [8]
Причины синдрома Беквита-Видеманна
Синдром Беквита-Видеманна со сложным типом наследования, локус заболевания расположен на коротком плече хромосомы 11 (CDKN1C, H19, IGF2, и KCNQ1OT1 гены). Аномальное метилирование нарушает регуляцию этих генов, что приводит к чрезмерно быстрому росту и другим характерным особенностей синдрома Беквит-Видемана.
Около 1% всех людей с данным синдромом имеют хромосомные аномалии, такие как перегруппировки (транслокации), ненормальное копирование (дублирование), или утраты (удаления) генетического материала из хромосомы 11.
Возможна молекулярно-генетическая верификация изменений этого локуса.
[9], [10], [11], [12], [13]
Симптомы синдрома Беквита-Видеманна
Заболевание характеризуется преждевременным быстрым ростом ребенка в раннем возрасте. После 8 лет рост замедляется. У некоторых детей с синдромом Беквит-Видемана отдельные части тела с одной стороны могут вырасти до аномально больших размеров (так называемая гемигиперплазия), что приводит к асимметричности внешнего вида.
Некоторые младенцы с синдромом Беквит-Видемана имеют аномально большой язык (макроглоссия), что иногда затрудняет дыхание и глотание, аномально большие органы брюшной полости (спланхномегалия), кожные складки или ямки возле ушей, гипогликемию и аномалии почек.
Дети имеют повышенный риск развития нескольких типов раковых опухолей, в частности, рака почки, опухоли Вильмса и гепатобластомы.
[14], [15], [16]
Осложнения и последствия
Возможные осложнения у больных с синдромом Беквита-Видеманна:
- вероятность неонатальной гипогликемии (60%) с развитием судорог, обусловленных транзиторным гиперинсулинизмом;
- высокая частота (10-40%) эмбриональных опухолей, особенно при нефромегалии или соматической асимметрии тела, требует наблюдения и проведения ультразвукового исследования почек 3 раза в год до 3-летнего возраста и в последующем 2 раза в год до 14-летнего возраста (своевременная диагностика опухоли Вильмса).
[17], [18], [19], [20], [21]
Диагностика синдрома Беквита-Видеманна
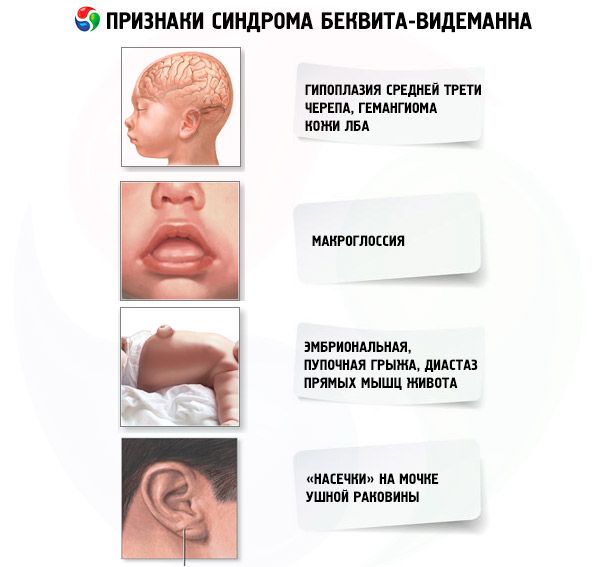
Диагноз синдром Беквита-Видеманна необходимо рассматривать у детей с аномалиями передней стенки живота (эмбриональной или пупочной грыжей, расхождением прямых мышц), макроглоссией, неонатальной гипогликемией и опухолями (нейробластомой, опухолью Вильмса, карциномой печени).
Диагностические критерии:
- Большая масса тела при рождении или постнатальное опережение физического развития.
- Дефекты закрытия передней стенки живота (эмбриональная, пупочная грыжа, диастаз прямых мышц живота).
- Висцеромегалия (нефромегалия, гепатомегалия, спленомегалия).
- Макроглоссия.
- Необычное лицо (гипоплазия средней трети, гемангиома кожи лба, «насечки» на мочке ушной раковины).
[22], [23], [24], [25], [26]
Какие анализы необходимы?
Лечение синдрома Беквита-Видеманна
Дефекты брюшной стенки устраняются с помощью хирургического лечения.
Гипогликемия у новорожденных с BWS должна лечиться в соответствии со стандартными протоколами терапии неонатальной гипогликемии.
Макроглоссия с возрастом часто становится менее заметной и не требует какого-либо лечения. В тяжелых случаях, макроглоссия устраняется с помощью операции. Некоторые хирурги рекомендуют выполнять оперативное вмешательство между 3 и 6 месяцами.
Гемигипертрофия в тяжелых случаях корректируется ортопедическими методами.
[27]
Прогноз
Синдром Беквита-Видеманна имеет разный прогноз для жизни. Он определяется своевременной диагностикой гипогликемии (профилактика умственной отсталости) и ранней диагностикой эмбриональных опухолей.
Продолжительность жизни, как правило, не отличается от здоровых людей.
Источник
Рубрика МКБ-10: Q87.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q80-Q89 Другие врожденные аномалии пороки развития / Q87 Другие уточненные синдромы врожденных аномалий пороков развития, затрагивающих несколько систем
Определение и общие сведения[править]
Синдром Беквита-Видемана
Генетическое заболевание, характеризующееся чрезмерно быстрым ростом, предрасположенностью к опухолям и врожденными пороками развития.
Распространенность: 1 — 5 / 10 000, наследование аутосомно-доминантное или возникает спорадически (85% случаев)
Этиология и патогенез[править]
Синдром Беквита-Видемана вызывается различными эпигенетическими и / или генетическими изменениями, которые приводят к дисрегуляции импринтированных генов на хромосоме 11p15.5.
Клинические проявления[править]
Пациенты демонстрируют ускоренный рост во второй половине беременности и в первые несколько лет жизни. Показатели роста взрослых пациентов обычно в пределах нормы. Аномальный рост может также проявляться в виде гемигиперплазии и/или макроглоссии (что приводит к трудностям при кормлении, затруднениям речи и нечасто к апноэ во сне). Гипогликемия наблюдается у 30-50% новорожденных. В дополнение к макросомии, макроглоссии, гемигиперплазии и гипогликемии характерными симптомами могут быть: омфалоцеле/пупочная грыжа/диастаз прямой мышцы живота, эмбриональная опухоль, передняя складка ушной раковины и задняя спиральная ямка, пламенеющий невус или другие сосудистые аномалии, висцеромегалия с участием органов брюшной полости, эмбриональная адренокортикальная цитомегалия (патогномоничный признак), аномалии почек и редко расщепление неба. Пороки сердца обнаруживаются в 9-34% случаев патологии, около половины из них составляет спонтанно разрешающаяся кардиомегалия. Кардиомиопатия встречается редко. Пациенты предрасположены к развитию эмбриональных злокачественных новообразований, главным образом, в первые 8 лет жизни с оценкой риска порядка 7,5% (диапазон 4-21%).
Синдромы врожденных аномалий, проявляющихся избыточным ростом (гигантизмом) на ранних этапах развития: Диагностика[править]
Диагностические признаки включают макроглоссию, макросомию и грыжу пупочного канатика.
Дифференциальный диагноз[править]
Дифференциальные диагнозы включают синдромы Симпсона-Голаби-Беммеля, Костелло, Перлмана и Сотоса, а также мукополисахаридоз VI типа.
Синдромы врожденных аномалий, проявляющихся избыточным ростом (гигантизмом) на ранних этапах развития: Лечение[править]
У новорожденного может возникнуть обструкция дыхательных путей. Попытки вентиляции при помощи дыхательного мешка через маску малоэффективны. Для облегчения самостоятельного дыхания ребенка кладут на бок. Можно использовать ротовой воздуховод (важно правильно подобрать размер). Поскольку интубация трахеи обычно затруднена, в экстренных случаях производят коникотомию.
Прогноз
При тяжелом варианте патологии пациенты подвержены риску ранней смерти из-за осложнений, возникающих вследствие гипогликемии, преждевременных родов, кардиомиопатии, макроглоссии или развитию опухоли. У пациентов, переживших детство, прогноз в целом хороший.
Профилактика[править]
Прочее[править]
Синдром Сотоса (церебральный гигантизм)
Синдром Сотоса является редким мультисистемным генетическим заболеванием, характеризуется типичной внешностью, чрезмерно быстрым ростом тела в раннем периоде жизни, макроцефалией и умственной отсталостью разной степени выраженности.
Изолированная гемигиперплазия
Синонимы: гемикорпоральная гипертрофия, изолированная гемигипертрофия
Изолированная гемигиперплазия — это редкий синдром чрезмерного роста, характеризующийся асимметричной локальной гипертрофией тела, затрагивающей по крайней мере одну конечность и сочетающийся с повышенным риском развития эмбриональных опухолей, главным образом нефробластомы и гепатобластомы.
Синдром Перлмана
Синонимы: нефробластоматоз-фетальный асцит-макросомия-опухоль Вильмса
Определение и общие сведения
Синдром Перлмана характеризуется главным образом полигидрамниозом, макросомией новорожденных, двусторонними почечными опухолями (гамартомами с нефробластоматозом или без него), гипертрофией островков Лангерганса и лицевым дисморфизмом.
До сих пор в литературе сообщалось о 30 пациентах с синдромом Перлмана. Представляется, что синдром Перлмана наследуется аутосомно-рецессивным способом.
Клинические проявления
Характерный лицевой дисморфизм включает в себя высокую линию волос головы спереди, низкую переносицу, гипотоничный внешний вид с открытым ртом, выступающая вывернутая верхняя губа и умеренная микрогнатия. Агенез мозолистого тела, гемангиомы сосудистого сплетения, расщепление неба, декстрапозиция сердца, прерывистая дуга аорты, диафрагмальная грыжа, висцеромагалия (включая нефромегалию, гепатомегалию, кардиомегалию, гиперплазия тимуса), фиброз печени с порто-портальным мостиком, абдоминальная мышечная гипоплазия, атрезия дистальной подвздошной кишки и крипторхизм — также были описаны у некоторых пациентов и, возможно, являются компонентом синдрома Перлмана. Гиперинсулинизм, по-видимому, является важной особенностью этого заболевания и может быть причиной смерти пациентов.
Диагностика
Пренатальная диагностика может быть ориентирована на проведение УЗИ, обнаржуние макроглоссии и почечных аномалий (кисты или гипертрофия).
Дифференциальный диагноз
Основными дифференциальными диагнозами являются синдромы Беквита-Видемана и Симпсона-Голаби-Бемеля: мутации гена GPC3 были исключены как причина синдрома Перлмана, генетические или эпигенетические изменения в области 11p15 никогда не обнаруживались у пациентов с синдромом Перлмана, несмотря на сильное фенотипическое сходство между двумя синдромами.
Лечение
Лечение поддерживающее и должно быть многопрофильным.
Прогноз
Прогноз синдрома Перлмана неудовлетворительный с высокой смертностью, особенно в неонатальном периоде из-за развития сепсиса или прогрессирующей респираторной недостаточности. Среди младенцев, выживших за пределами неонатального периода, две трети обнаруживают опухоль Вильмса и большинство из них имели некоторую степень задержки развития. Тем не менее, одна девочка, как сообщается, была жива в возрасте 9 лет и показала нормальное психомоторное развитие.
Синдром Симпсона-Голаби-Бемеля
Определение и общие сведения
Синдром Симпсона-Голаби-Бемеля представляет собой редкий синдром X-сцепленных множественных врожденных аномалий, характеризующийся пре- и постнатальным гигантизмом, характерными черепно-лицевыми особенностями, разнообразными врожденными аномалиями, органомегалией и повышенным риском развития опухолей.
Распространенность неизвестна. До настоящего времени было зарегистрировано около 250 случаев синдрома Симпсона-Голаби-Бемеля.
Этиология и патогенез
Синдром Симпсона-Голаби-Бемеля возникает из-за мутаций с потерей функции в гене GPC3 (Xq26), кодирующим Glypican-3 (GPC3) — протеогликан сульфата гепарина на поверхности клетки, который действует как отрицательный регулятор сигнального пути Hedgehog. Мутации в гене GPC3 приводят к гиперактивации сигнального пути Hedgehog, что в конечном итоге приводит к гигантизму и малигнизации. Дупликация гена GPC4 была также идентифицирована в одном случае заболевания.
Клинические проявления
Синдром Симпсона-Голаби-Бемеля имеет широкую клиническую картину с различной степенью тяжести. Характеризуется пре- и постнатальным гигантизмом с макросомией, характерными черепно-лицевыми особенностями (макроцефалия с грубыми чертами лица, макроглоссия, гипертелоризм, зубочелюстная недостаточность, небные аномалии), добавочные соски, врожденные пороки сердца и аритмия, сегментные дефекты позвонков, висцеромегалия (почечная дисплазия/нефромегалия, спленомегалия и гепатомегалия), диафрагмальная грыжа, диастаз прямой мышцы живота/пупочная грыжа, аномалии конечностей (полидактилия/брахидактилия, кожная синдактилия, гипоплазия ногтей) и участие крипторхизм, гипоспадия. Вовлечение центральной нервной системы включает в себя различную степень умственной неполноценности, задержки двигательного и речевого развития. Пациенты с синдромом Симпсона-Голаби-Бемеля подвергаются повышенному риску возникновения эмбриональных опухолей (опухоль Вильмса), гепатобластома, надпочечниковая нейробластома, гонадобластома, гепатоцеллюлярная карцинома). Также была описана летальная форма синдрома, известная как синдром Симпсона-Голаби-Бемеля 2-го типа, характеризующаяся развитием водянки плода.
Синдром Протея
Синонимы: парциальный гигантизм-невус-гемигипертрофия-макроцефалия синдром
Определение и общие сведения
Синдром Протея — очень редкое и комплексное гамартаматозное заболевание, характеризующееся прогрессирующей гипертрофией скелета, кожи, жировой ткани и центральной нервной систем.
До настоящего времени было зарегистрировано около 120 случаев синдрома Протея. Предполагается, что распространенность составляет менее 1/1 000 000 живорождений.
Этиология и патогенез
Кузальные мутации при синдроме Протея идентифицированы в двух компонентах сигнального пути фосфатидилинозитол-3-киназы-AKT: гена PTEN, которые делают синдром Протея частью синдрома опухоли PTEN гамартома и гена AKT1. Мутация гена AKT1 представляет собой случай соматического мозаицизма, который, происходит в 1-47% случаев. Мутации PTEN затрагивают последовательнсть ДНК, а также присутствуют также в виде соматического мозаицизма.
Клинические проявления
Новорожденные обычно нормальны при рождении. Манифестация синдрома Протея обычно происходит в 6-18 месяцев асимметричным увеличением главным образом рук и ног. Макродактилия является наиболее распространенным симптомом синдрома Протея, наряду с гемигипертрофией. Гипертрофия скелета может быть выраженной и быстро прогрессирующей с развитием причудливых, уродующих, нерегулярных кальцинированных разрастаний трубчатых костей конечностей, черепа и тел позвонков. Церебриформные соединительнотканные невусы могут возникать на любом участке тела и обычно развиваются позже в детстве. Сосудистые мальформации и линейные эпидермальные невусы наблюдаются в первые месяцы жизни и в целом стабилизируются со временем. В младенчестве отмечаются нарушения жировой ткани и сосудистые мальформации. Неврологические проявления включают интеллектуальный дефицит, тромбоз синуса и внутричерепные поражения. Осложнения синдрома Протея включают гемимегалэнцефалию, буллезную болезнь легких, легочную эмболию и тромбоз глубоких вен. Сообщалось о случаях в основном доброкачественных опухолей, редко злокачественных, таких как как папиллярная аденокарцинома яичка, менингиома и цистаденома яичников.
Диагностика
Диагноз синдрома Протея базируется на выполнении клинических критериев. Основные критерии — мозаичное распределение поражений, спорадическое возникновение, прогрессивное течение, должны сопровождаться наличием церебриформных соединительнотканных невусов (категория А) или с 2-мя критериями категории B (асимметричный непропорциональный рост, линейный эпидермальный невус, специфические опухоли до 2-го десятилетия жизни) или 3-мя признаками категории С (сосудистые мальформации, нарушения жировой ткани, характерный фенотип лица). Если критерии не соблюдаются, пациенту может быть поставлен диагноз с протеоподобного синдрома. Молекулярно-генетическое тестирование может подтвердить диагноз синдрома Протея.
Дифференциальный диагноз
Дифференциальный диагноз синдрома Протея включает синдром Клиппеля-Треноне-Вебера, гемигипертрофию, болезнь Оллье, макродактилию, синдром Маффуччи, синдром CLOVES, нейрофиброматоз тип 1 и другие синдрома опухоли PTEN гамартома.
Лечение
Лечение синдрома Протея требует многодисциплинарного подхода. Вмешательствами, используемыми для контроля над гипертрофией трубчатых костей, являются эпифизиостаз, эпифизидезис и в крайних случаях ампутация. Очень важна физическая и профессиональная терапия. Может потребоваться специальная ортопедия или обувь. Повреждения кожи следует удалять хирургическим путем только, если подозревается злокачественность или если присутствует значительная деформация и/или боль. Если развивается легочная эмболия и тромбоз глубоких вен, следует незамедлительно следовать начать прием антикоагулянтов. Пациентов следует регулярно обследовать на наличие опухолей. Психосоциальное консультирование может быть полезным для пациентов и их семей. Также рекомендуется ежегодный физикальный осмотр и рентгенография.
Прогноз
Прогноз синдрома Протея зависит от тяжести осложнений.
Гигантизм стоп у детей
Клиническая картина
В зависимости от вида деформации выделено пять вариантов гигантизма стоп у детей: гигантизм всей стопы, внутреннего, среднего, наружного её отделов и макродактилия.
Лечение
Лечение деформаций нижних конечностей у больных гигантизмом — крайне сложная и мало разработанная проблема.
а) Консервативное лечение
Консервативное лечение врождённого гигантизма стоп у детей неэффективно.
б) Хирургическое лечение
Используют различные способы в зависимости от вида деформации. Оптимальный возраст для операции — 6 мес.
При тотальном увеличении всей стопы у детей младшего возраста показаны следующие вмешательства: эпифизиодез ростковых зон плюсневых костей в сочетании с их периостэктомией и иссечением межкостных мышц для устранения мягкотканых препятствий к сужению поперечного свода стопы. Сближенные в результате лучи фиксируют сухожильным аутотрансплантатом, выкроенным из длинных разгибателей II-III пальцев, который огибает плюсневые кости в виде восьмёрки, и прочно фиксируют капроновой нитью.
При тотальном увеличении всей стопы, когда она достигает обезображивающего размера, показана вынужденная экзартикуляция одного-двух наиболее увеличенных средних лучей с клиновидной резекцией костей предплюсны. Фиксацию осуществляют путём остеосинтеза аутотрансплантатом, внедрённым в соседние плюсневые кости, и спицами Киршнера. Производят эпифизиодез оставшихся плюсневых костей и фаланг пальцев, дефатизацию и кожную пластику.
При изолированном увеличении одного или нескольких лучей внутреннего, среднего или наружного отделов выполняют многоэтапные операции. Первым этапом производят экзартикуляцию одного из наиболее увеличенных лучей с клиновидной резекцией костей стопы на уровне среднего отдела. Вторым и последующим этапами выступают укорачивающие моделирующие резекции фаланг пальцев и плюсневых костей, направленные на уменьшение продольного размера стопы,
а также продольные резекции фаланг пальцев и плюсневых костей, направленные на уменьшение поперечного размера стопы.
Источники (ссылки)[править]
Педиатрия [Электронный ресурс] / Под ред. А.А. Баранова — М. : ГЭОТАР-Медиа, 2009. — https://www.rosmedlib.ru/book/ISBN9785970410851.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
1. American Academy of Pediatrics and The American College of Obstetricians and Gynecologists. Guidelines for Perinatal Care. Chicago, IL, 1992.
2. Fanaroff M., Martin R. J. Neonatal-Perinatal Medicine: Diseases of the Fetus and Newborn. St. Louis: Mosby-Year Book, 1992.
3. Fletcher M. A., MacDonald M. G. Atlas of Procedures in Neonatology. Philadelphia: Lippincott, 1993.
4. Jones K. L. Smith’s Recognizable Patterns of Human Malformations. Philadelphia: Saunders, 1988.
5. Taeusch H. W., Ballard R. A., Avery M. E. Schaeffer and Avery’s Diseases of the Newborn. Philadelphia: Saunders, 1991.
Действующие вещества[править]
Источник