Синдром арнольда киари фото узи

Общая информация
Синдром Арнольда-Киари представляет собой набор признаков и симптомов, вызванных редкой мальформацией (отклонение от нормального развития, аномалия) задней черепной ямки; у пострадавших эта структура развита слабо, поэтому мозжечок выходит (выступает) из своего естественного участка через затылочное отверстие, расположенное у основания черепа.
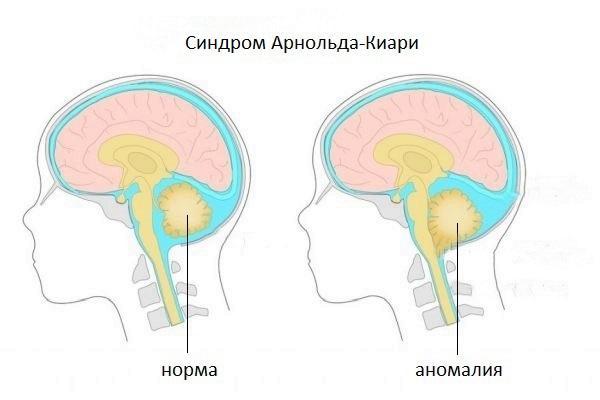
Есть четыре различных типа синдрома Арнольда-Киари; особенность, отличающая один тип от другого, является степень выпячивания, следовательно, доля вовлеченного материала мозжечка. Тип I является наименее тяжелым (иногда остается бессимптомным на протяжении всей жизни), тогда как IV тип наиболее тяжелый; однако уже со второго типа качество жизни больного ставится под угрозу.
Симптомы, характеризующие аномалии Арнольда-Киари многочисленны и варьируются от головных болей до слабости мышц и проч.
На сегодняшний день не существует лекарств, позволяющих устранить порок развития мозжечка, однако существуют способы лечения, позволяющие частично смягчить симптомы.
Что такое синдром Арнольда-Киари?
Синдром Арнольда-Киари, или мальформация Арнольда-Киари — структурное изменение мозжечка, характеризующееся его смещением вниз, именно в направлении позвоночного канала и затылочного отверстия, базальные части полушария мозжечка.
Простыми словами, это грыжа мозжечка, при которой часть мозжечка выступает из затылочного отверстия, проникая в позвоночный канал.
Синдром Арнольда-Киари получил свое название от двух врачей, которые впервые описали его, Арнольда Джулиуса и Ганса Киари.
Причины и факторы риска
Исследователи полагают, что синдром Арнольда-Киари может иметь наследственное происхождение, так как обнаруживалась среди членов одной семьи. Тем не менее, генетические условия, вызывающие заболевание (т.е., какие и сколько генов участвуют) и тип передачи еще предстоит выяснить.
Исходя из серьезности выпячивания и момента жизни, в котором он возникает, заболевание можно разделить на 4 различных типа, идентифицированных первыми четырьмя римскими числами (I, II, III и IV).
Первые два типа по сравнению со вторыми более распространены и менее серьезны; Тип III и тип IV, на самом деле, очень редки и несовместимы с жизнью.
— Мальформация I типа.
Первая степень синдрома протекает бессимптомно (т.е. без явных симптомов), по крайней мере, до конца детства или юности.
Причина его возникновения кроется в уменьшенном черепном пространстве: в таких условиях часть мозжечка (именно миндалина(и), расположенная(ые) с нижней стороны), из-за недостатка места, вынуждена проникать в затылочное отверстие и входить в позвоночный канал.
Примечание: у некоторых взрослых людей с синдром Арнольда-Киари 1 типа все в порядке и они ведут совершенно нормальную жизнь. Это связано с тем, что аномалия мозжечка не настолько серьезна, чтобы вызывать симптомы или нарушения. Поэтому очень часто эти субъекты игнорируют свое состояние или узнают о нем по чистой случайности.
— Мальформация II типа.
2 тип мальформации Арнольд-Киари является врожденным заболеванием, которое присутствует с рождения ребенка, и всегда протекает симптоматически.
По сравнению с 1 степенью он характеризуется большим выпячиванием черепной ямки, при котором помимо миндалин мозжечка также выпячивает часть мозжечка (называемая червь мозжечка) и венозный сосуд.
Почти всегда мальформация Арнольд-Киари II типа ассоциируется с особой формой расщелины позвоночника, называемой миеломенингоцеле.
Среди различных последствий этой аномалии выделают: блокирование потока ликвора (спинномозговой жидкости) через затылочное отверстие (что приводит к состоянию, называемому гидроцефалией) и прерывание нервных сигналов.
Первоначально термин Арнольд-Киари относился только ко 2 типу заболевания. Теперь, её обычно используют для всех форм болезни.
— Мальформация III типа.
Присутствующий с рождения, III тип порока вызывает серьезные неврологические проблемы, настолько, что часто несовместимы с жизнью. В этих случаях на самом деле наблюдается выпячивание мозжечка, и по этой причине говорится о затылочном энцефалоцеле.
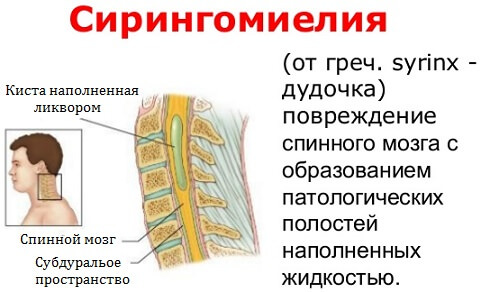
Обычно III тип состояния характеризуется гидроцефалией и сирингомиелией; последний представляет собой особое состояние, характеризующееся наличием одной или нескольких кист в позвоночном канале.
— Мальформация IV типа.
Мальформация Арнольда-Киари IV типа характеризуется отсутствием развития части мозжечка (недоразвитие мозжечка).
Аномалия врожденная и абсолютно несовместима с жизнью.
Связанные расстройства
Врачи и ученые отметили, что следующие заболевания являются частыми среди людей с мальформацией Киари:
- гидроцефалия;
- сирингомиелия;
- сколиоз;
- синдром Марфана;
- синдром Элерса-Данлоса.
Эпидемиология
Точная частота пороков развития неизвестна; это связано с тем, что у некоторых даже взрослых людей с I типом мальформации Арнольда-Киари никаких симптомов нет, и они кажутся совершенно нормальными (поэтому болезнь недиагностируется).
Несколько достоверных эпидемиологических исследований сообщают, что:
- I тип симптоматичен у 1 из 100 детей;
- II тип особенно широко распространен в популяциях кельтского происхождения;
- женщины страдают в 3 раза чаще, чем мужчины.
Симптомы и осложнения

4 типа заболевания имеют разные симптомы и признаки.
Ниже приводится таблица с точным описанием симптомов, которые характеризуют I, II и III типы синдрома.
Для IV типа невозможно проследить симптоматику, так как это состояние неизбежно и внезапно приводит к гибели плода.
| Мальформация I типа | Мальформация II типа | Мальформация III типа |
Когда у больного 1 стадия, симптомы следующие:
| Синдром Арнольда-Киари II типа характеризуется теми же симптомами, что и тип I, с той разницей, что они имеют более выраженную интенсивность и присутствуют всегда. Кроме того, если он сопровождается миеломенингоцеле (см. ниже), II тип состояния также вызывает:
| Люди с мальформацией III типа страдают от серьезных неврологических проблем (часто несовместимых с нормальной жизнью), гидроцефалии и сирингомиелии. Последний, характеризуется образованием одной или нескольких кист внутри спинного мозга, и может вызывать:
|
Расщелина позвоночника (миеломенингоцеле).
Расщелина позвоночника — врожденный порок развития позвоночника, из-за которого менинги, а иногда и спинной мозг выходят из своего места (обычно они ограничены позвонками). Миеломенингоцеле является наиболее тяжелой формой расщелины позвоночника: у пораженных выпячивают менинги и спинной мозг из позвоночной камеры и образуют мешок на уровне спины. Эта сумка, хотя и защищена слоем кожи, подвержена внешним воздействиям и постоянно подвергается риску серьезных, а в некоторых случаях даже смертельных инфекций.
Синдром Арнольда-Киари типа II, III и IV видны уже в пренатальном возрасте (т.е., когда пораженный ребенок все еще находится в материнской утробе) при ультразвуковом исследовании.
Что касается I типа, желательно обратиться к врачу, как только появятся типичные симптомы, о которых говорилось выше. Важно также проходить своевременные обследования, так как в результате последних могут возникнуть другие сопутствующие нарушения.
Осложнения
Осложнения синдром Арнольда-Киари связаны с ухудшением выпячивания мозжечка или патологическими состояниями, связанными, следовательно, с гидроцефалией, миеломенингоцеле, сирингомиелией и проч.
Ухудшение выпячивания (протрузии), обусловленное повышенным давлением черепа на мозжечок, очевидно, предполагает обострение симптомов.
Диагностика
Диагностические тесты, которые позволяют установить степень протрузии мозжечка через затылочное отверстие (таким образом, устанавливая тип мальформации Арнольда-Киари):
- Магнитно-резонансная томография (МРТ). Благодаря формированию магнитных полей, он позволяет получить детальное изображение мозжечка и позвоночного канала, не подвергая пациента вредному ионизирующему излучению.
- Компьютерная томография (КТ) дает четкие изображения внутренних органов, в том числе мозжечка и спинного мозга. Во время его выполнения субъект подвергается минимальному воздействию вредного ионизирующего излучения.
КТ и МРТ, которым предшествует точное физическое обследование, имеют основополагающее значение для выявления любых патологий, связанных с синдромом Арнольда-Киари.
Таблица. Как и когда диагностируется аномалия Арнольда-Киари развития заболевания.
| Тип порока развития | Когда и как это можно диагностировать? |
| I | В позднем детстве или позднем подростковом возрасте посредством объективных обследований с последующим КТ и/МРТ. |
| II | В дородовом возрасте при УЗИ. При рождении и в раннем детстве, посредством объективных обследований, компьютерной томографией и/или МРТ. |
| III | В дородовом возрасте при УЗИ. После рождения и в раннем детстве, посредством объективных обследований, компьютерной томографией и/или МРТ. |
| IV | В дородовом возрасте при УЗИ. |
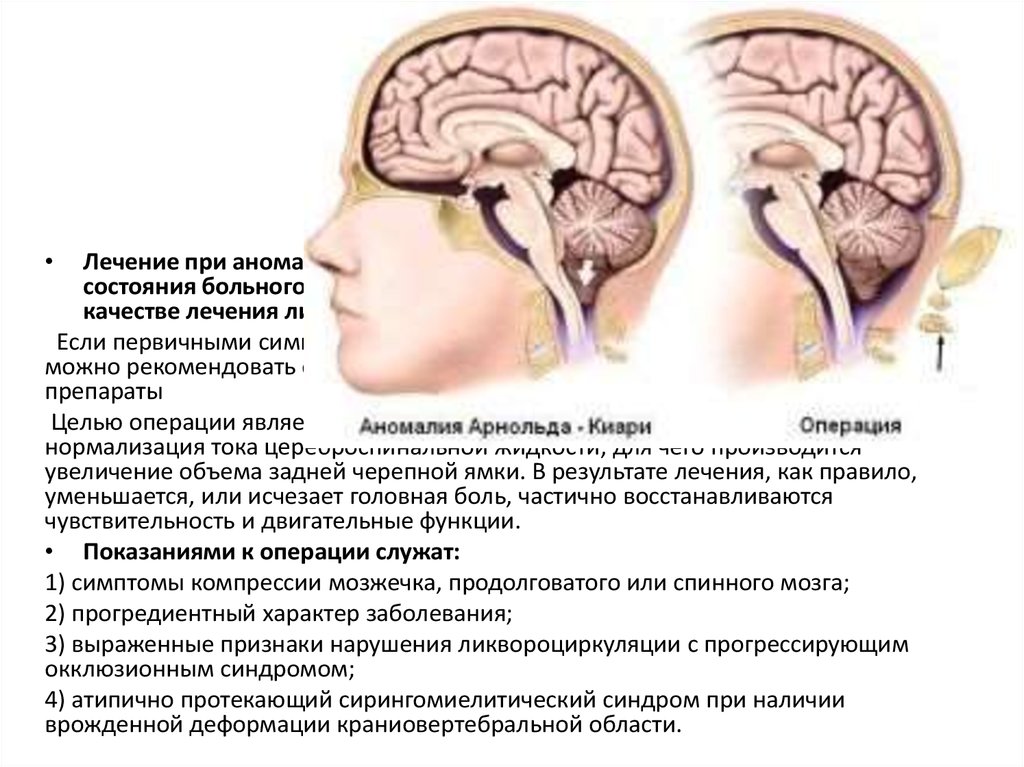
Лечение
Синдром Арнольда-Киари неизлечим. Однако существуют как фармакологические, так и хирургические методы терапии, позволяющие частично смягчить признаки заболевания.
— Медикаментозная терапия.
Пациенты с мальформацией Арнольда-Киари I типа, страдающий от головной боли и боли в шее и/или лице, могут принимать обезболивающие препараты.
Выбор наиболее подходящих лекарств для конкретного случая остается за лечащим врачом.
— Хирургическое лечение.
Цель хирургического лечения состоит в том, чтобы уменьшить давление, оказываемое черепом, чтобы предотвратить повреждение мозжечка и спинного мозга.
Для достижения этой цели есть несколько процедур, таких как:
- Декомпрессия задней черепной ямки, во время которой хирург удаляет часть задней части затылочной кости.
- Декомпрессия спинного мозга с помощью ламинэктомии (декомпрессионная ламинэктомия). Во время его выполнения хирург удаляет пластинку второго и третьего шейного позвонка. Пластинка — это позвоночная часть, отделяющая отверстие, через которое проходит спинной мозг..
Примечание: иногда декомпрессия задней ямки и декомпрессивная ламинэктомия выполняются одновременно. - Декомпрессионный разрез твердой мозговой оболочки. При разрезе твердой мозговой оболочки или наружного менинга пространство, доступное мозжечку, увеличивается, а давление на его повреждение уменьшается. Чтобы покрыть и защитить трещину, созданную разрезом, хирург пришивает на нее кусок искусственной ткани (или взятый из другой части тела).
- Хирургическое шунтирование (создание дополнительного пути в обход пораженного участка). Это, по сути, дренажная система, состоящая из гибкой трубки, позволяющей удалять спинномозговую жидкость, в случае гидроцефалии, или опорожнять цисту(ы), в случае сирингомиелии. Не исключено, что пациенты с гидроцефалией будут вынуждены проходить хирургическим шунтом всю жизнь.
— Осложнения хирургического вмешательства.
Риски, связанные с хирургией, различны. Фактически возможно появление:
- кровотечений;
- повреждение структур головного мозга и/или спинного мозга;
- инфекционный менингит;
- проблемы с заживлением ран;
- необычные скопления жидкости вокруг мозжечка.
Помните, что любое повреждение головного или спинного мозга, произошедшее во время операции, непоправимо. Поэтому, прежде чем подвергнуться больного вмешательству любого типа, лечащий врач выявит любые риски и осложнения необходимой процедуры.
Прогноз
Синдром Арнольда-Киари типа II, III и IV никогда не имеют положительного прогноза, поскольку, помимо того, что неизлечимы, могут вызывать серьезные неврологические нарушения или даже быть несовместимыми с жизнью.
Прогноз для пациентов с I типом часто неизвестен. Многие люди с этим заболеванием не имеют никаких симптомов, и невозможно предсказать, будут ли симптомы развиваться в будущем. Другие люди с мальформацией Арнольда-Киари могут испытывать головокружение, мышечную слабость, онемение, проблемы со зрением, головную боль или проблемы с равновесием и координацией. У этих людей не всегда возможно предсказать, будут ли симптомы ухудшаться с течением времени.
Людям с пороком 1 типа важно регулярно проходить медицинские обследования, чтобы быть под наблюдением врача при появлении любых новых симптомов.
Источник
Синдром Арнольда-Киари — это порок развития, относится к так называемым дефектам развития нервной трубки плода, встречается примерно 3-8 случаев на 100000 новорожденных. На вопросы пациентов о синдроме Арнольда-Киари отвечают врачи медицинских клиник «Арт-Мед».
Мне 26 лет. МРТ диагностика показала, что у меня синдром Арнольда-Киари 1 степени. Можно ли вылечиться, или необходимо с этим смириться? Как в последствии этот диагноз отразиться на моем здоровье? Могу ли я рожать, если да то будет ли этот диагноз у моего ребенка?
Синдром Арнольда-Киари относится к врожденным аномалиям строения костей черепа в области сочленения головного и спинного мозга; при данной патологии может происходить опущение части мозжечка в большое затылочное отверстие. В зависимости от тяжести клинических проявлений (головные боли, боль в шее, руке) решается вопрос о необходимости хирургического лечении. Обычно при I степени аномалии клинические проявления бывают нетяжелыми, поэтому и беременность, как правило, возможна. Такие пациенты обычно находятся под наблюдением невропатолога, нейрохирурга. Риск наследования плодом подобной аномалии существует, но не более 5 — 10%. Во время беременности тяжелые формы синдрома Арнольда-Киари хорошо диагностируются у плода с помощью УЗИ.
Мне 28 лет, обнаружен синдром Арнольда-Киари. Можно ли излечиться от этого диагноза, и каким методом?
Синдром Арнольда-Киари относится к врожденным особенностям строения костей черепа в области сочленения головного и спинного мозга; при данной патологии может происходить опущение части мозжечка в большое затылочное отверстие. В зависимости от тяжести клинических проявлений (головные боли, боль в шее, руке) решается вопрос о необходимости хирургического лечении. Такие пациенты обычно находятся под наблюдением невролога, нейрохирурга.
Мне 36 лет, у меня двое детей (16 и 9 лет). Сейчас в повторном браке, мужу 32 года. Хотим родить здорового ребенка, в 2007 году забеременела, но в 20 недель пришлось прерывать беременность (синдром Арнольда-Киари и гидроцефалия плода). Причину возникновения врачи нам не объяснили, сказали, что произошел хромосомный надлом, и можно спокойно беременеть через полгода. Какие причины возникновения такого порока, что надо делать для того, чтобы родить здорового ребенка? Или мой возраст дает большой риск?
В Вашем случае у детей имелись так называемые мультифакториальные (=многофакторные) аномалии развития центральной нервной системы (ЦНС), указать конкретную причину формирования данных аномалий, как правило, невозможно. Риск повторения существует, учитывая два случая проблем развития ЦНС у детей, повторный риск оценила бы не менее 8-10% (средний генетический риск). Принципиальных возражений против планирования беременности в Вашем браке (на основании этой информации), конечно же, нет. Существенную роль в формировании дефектов ЦНС плода играет дефицит фолиевой кислоты на самых ранних сроках. Поэтому Вам и рекомендован прием этого препарат в качестве подготовки к беременности. Советую начать прием препаратов фолиевой кислоты с момента отмены контрацепции (= предохранения от беременности). Ваш возраст 36 лет вряд ли имеет отношение к формированию пороков ЦНС у плода, но может немного повышать риск рождения ребенка с другим видом генетической патологии – хромосомными заболеваниями, в частности, с синдромом Дауна. Поэтому все беременные старше 35 лет должны находиться под наблюдением генетика с 10-12 недель, им предлагается дополнительный комплекс пренатальной диагностики. Более подробно все эти вопросы можно обсудить на очной генетической консультации в нашем медицинском центре.
Источник
Врачи выделяют три вида аномалии Киари. Они зависят от изменений в структуре тканей мозга, проникающей в позвоночный отдел, а также от вида нарушений в работе и функционировании мозга спинного и головного.
Обычно симптомов этого заболевания нет, так что и лечить его не надо. Часто этот синдром обнаруживается только при исследовании других болезней.
Несмотря на кажущуюся безобидность, последствия аномалии Арнольда Киари могут быть плачевными.
Что это за явление
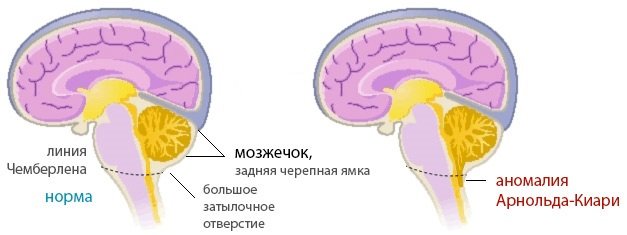
Мальформация Арнольда Киари встречается редко. При этом заболевании задняя часть мозга перемещается в каудальное направление, выпадает в затылочное отверстие.
В этом месте человек может ощущать тянущую боль, иногда могут возникать неврологические отклонения. Решение этой проблемы одно – операция, обычно выполняют шунтирование или декомпрессию ямки.
Классификация аномалии
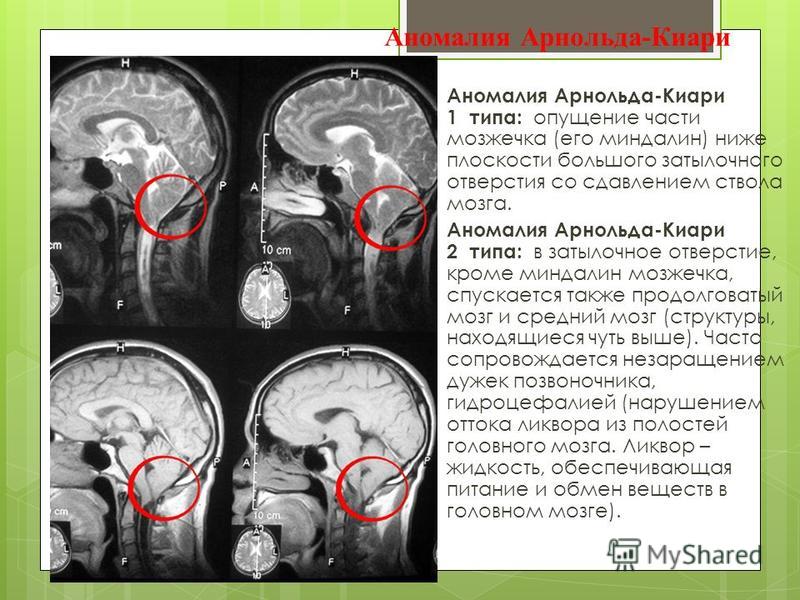
Аномалия Арнольда Киари иметь разную тяжесть, поэтому есть четыре вида аномалии.
- Для первого типа характерно опущение миндалин мозжечка, они находятся ниже отверстия в затылке. Обнаружить это можно у подростка или взрослого человека. При такой стадии может быть выявлена гидромиелия, когда в центральном канале спинного мозга оказывается жидкость.
- Для второго типа характерно проявление сразу же с рождения ребенка. Ее можно заподозрить, если через затылок выходят миндалины мозжечка и продолговатый мозг. При таком заболевании всегда есть жидкость в спинном мозге, а иногда еще может быть врожденная спинномозговая грыжа.
- Для третьего типа характерно особое положение мозжечка и продолговатого мозга в менингоцеле шейно-затылочного отдела.
- У четвертого типа диагностируется почти полное отсутствие мозжечка. Причем он не опускается. Это состояние еще называют синдромом Денди-Уокера, когда к этой патологии прибавляется гидроцефалия, сирингомиелия, иногда могут появляться кисты на задней части черепа.
При втором и третьем типе аномалии Арнольда Киари могут быть другие симптомы со стороны нервной системы:
- полимикрогирия;
- гетеротопия мозга;
- кисты в отверстии Можанди;
- изменение мозолистого тела;
- изменений работы подкорки;
- изменение строения сильвиевого водопровода;
- может появиться намет и серп мозжечка.
Симптоматика
Типичными признаками аномалии Арнольда Киари являются:
- Частые боли в районе затылка, которая становится сильнее в моменты кашля и чихания.
- Боли в голове из-за повышения давления или напряжения шеи.
- Головокружения, потеря сознания, если резко повернуть голову или встать.
- Падение зрения.
- Плохое самочувствие.
- Температура тела ниже нормы, боли в руках.
- Слабость в мышцах рук.
- Спастичность пальцев.
- Наблюдается апноэ.
- Становится трудно глотать.
- Мозг перестает контролировать движения зрачков.
- Проблемы с мочеиспусканием.
- Подергивания глаз.
- Шум в ушах, если наклониться резко или повернуться.
- Тремор рук и ног.
- Проблемы с координацией.
- Проблемы в развитии мелкой моторики.
- Потеря чувствительности тела.
- Слабеют мышцы, из-за чего становится сложно ходить или поднимать вещи.
- Инфаркт мозга (спинного или головного).
Причины появления

До сих пор нет точного ответа, откуда пришло это заболевание. Некоторые неврологи говорят, что дело в неправильном размере черепа человека, из-за того, что в ней мало места, мозг не вмещается, и его части выходят за пределы черепной коробки.
Также есть вариант, что это явление вызвано слишком большим размером мозга, за которым не успевает расти голова, поэтому орган вынужден размещаться иным способом, перемещая мозжечок за пределы черепа.
Есть еще одна версия аномалии Арнольда Киари. При незначительной выраженности заболевания долгое время оно может резко обостриться из-за гидроцефалии. Жидкость влияет на размер желудочков мозжечка, увеличивая размер этого отдела мозга.
Мальформация не дает нормально развиваться связочному аппарату затылочно-шейной части тела. Поэтому при ударе или другой травме мозг страдает еще сильнее, появляются симптоматика манифестации.
Если во время беременности у женщины была эктопия шейки матки, то есть большая вероятность того, что у ребенка будет обнаружен этот синдром. Если обнаружить это на ранних сроках, можно прервать беременность. Другие причины пока не выявлены.
Осложнения
У людей с синдромом Арнольда Киари может долго не наблюдаться серьезных патологий, но если заболевание начнет развиваться, это может привести с тяжелым последствиям.
Среди них:
- скопление жидкости в черепной коробке вокруг мозга;
- развитие паралича, если жидкость соберется вокруг спинного мозга, причем не всегда в этой ситуации помогает операция;
- сирингомиелия – киста в позвоночнике, где есть жидкость. Это образование давит на спинной мозг, нарушая его работу;
- пороки сердца наблюдаются редко;
- проблемы с дыханием;
- пневмония из-за застоя;
- низкий уровень интеллекта.
Диагностика
 Для постановки диагноза надо знать степень развития заболевания, чтобы скорректировать тактику лечения. Также важен очный осмотр и несколько процедур: эхо- и электро- ЭГ мозга, рэоэнцефалография.
Для постановки диагноза надо знать степень развития заболевания, чтобы скорректировать тактику лечения. Также важен очный осмотр и несколько процедур: эхо- и электро- ЭГ мозга, рэоэнцефалография.
Но их результаты не могут досконально подтвердить диагноз, потому что они показывают только уровень внутричерепного давления.
Рентген черепа нужен, чтобы выявить аномалии в строении костей, которые характерны для этого заболевания.
КТ и МСКТ мозга не дают возможность врачам хорошо рассмотреть участок краниовертебрального перехода, а также не показывают состояние мягкого вещества мозга в задней части черепа.
Поэтому для диагностики аномалии Арнольда Киари используется магнитно-резонансная томография, с помощью которой можно увидеть состояние и головного, и спинного мозга.
Если надо обследовать ребенка, ему введут успокоительное, потому что в процессе процедуры нужна полная неподвижность.
Кроме МРТ мозга, может понадобиться исследование области шеи и грудной клетки. Это бывает в том случае, если есть вероятность обнаружения кист на позвоночнике, давящих на спинной мозг.
МРТ является лучшим выбором и потому, что может показать незначительные отклонения в работе нервной системы, которые укажут на присутствие болезни.
Лечение
Использование медикаментов при аномалии Арнольда Киари оправдано только в том случае, если пациент жалуется только на боль в затылке или шее. Разработаны препараты, которые снимают воспаление и обезболивают.
Если же это не помогает, а болезнь прогрессирует, придется соглашаться на операцию. Хирурги устранят все возможные проявления аномалии, которые давят на мозг, могут восстановить циркуляцию ликвора.
Обычно есть два варианта операций:
- рассечение концевой нити;
- декомпрессия отверстия в затылке, или краниоктомия.
Рассечение концевой нити
Плюсы такой тактики лечения аномалии Арнольда Киари операции такие:
- Работа хирурга длится всего 45 минут. Все действия не требуют серьезного инвазивного вмешательства, при этом можно вернуть на место все части мозга.
- После операции восстановление происходит быстро, реабилитация не нужна.
- После процедуры исключается смерть человека от этой аномалии.
- Устраняются не только последствия, но и причины появления мальформации, а также ряда других заболеваний.
- Смертность от такой процедуры нулевая.
- После операции не начнется гидроцефалиия из-за смещения миндалины.
- Человек чувствует себя лучше, а болезнь не развивается.
- Начинает лучше работать кровообращение, нервная система восстанавливает свои функции.
- Возможность жить полной жизнью.
Есть и некоторые минусы у этой операции:
- Небольшой шрам на копчике.
- После операции место разреза будет болеть несколько дней.
- Из-за ухода спастичности кажется, что стало меньше силы в конечностях.
- Из-за улучшения кровоснабжения мозга может увеличиться активность этого органа.
- Во время восстановления человек может чувствовать себя некомфортно, но это состояние быстро проходит.
Краниотомия
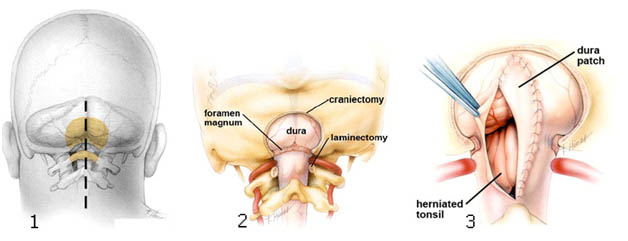
Схема оперативного лечения аномалии Арнольда Киари
Плюсы такой операции по декомпрессии затылка:
- Результат виден спустя пару дней после процедуры.
- Точно не наступит быстрая смерть от этого заболевания.
Но недостатков у декомпрессии при аномалии Арнольда Киари намного больше:
- Причина аномалии не исчезает.
- Смертность может достигать трех процентов.
- Работа хирурга оказывает сильное воздействие на организм, пациент может стать инвалидом.
- Улучшение есть, но не такое большое, как при первом методе лечения.
- Может развиться отек мозга.
- Возможно появление пневмоэнцефалии.
- После операции возможно появление гидроцефалии.
- Тетрапарез рук и ног.
- Во время процедуры может уйти порядка 14% от объема спинномозговой жидкости.
- Эмболия.
- Неврологический дефицит в редких случаях, зависит от зоны вмешательства.
- Появление инфекции в черепной коробке, из-за которой начнется менингит или появится абсцесс в мозге.
- Гемодинамические изменения ствола мозга.
- Появление эпидуральной гематомы.
- Кровоизлияние в мозге.
- Интрааксиальное кровоизлияние, из-за которого начинается неврологический дефицит.
Перед любым видом операции врачи тщательно обследуют пациента, чтобы понять, какой вариант будет более актуален в данном случае, а также изучают особенности организма, которые могут стать противопоказанием для хирургии.
Люди с такой аномалией могут жить долго, но не наслаждаться жизнью сполна. Поэтому операции показаны, чтобы дать возможность прочувствовать все прелести жизни без ограничений.
У всех методов лечения есть свои показания, поэтому всегда выбирается тот метод, который меньше всего навредит человеку.
Источник