Синдром арнольда киари 2 тип что это

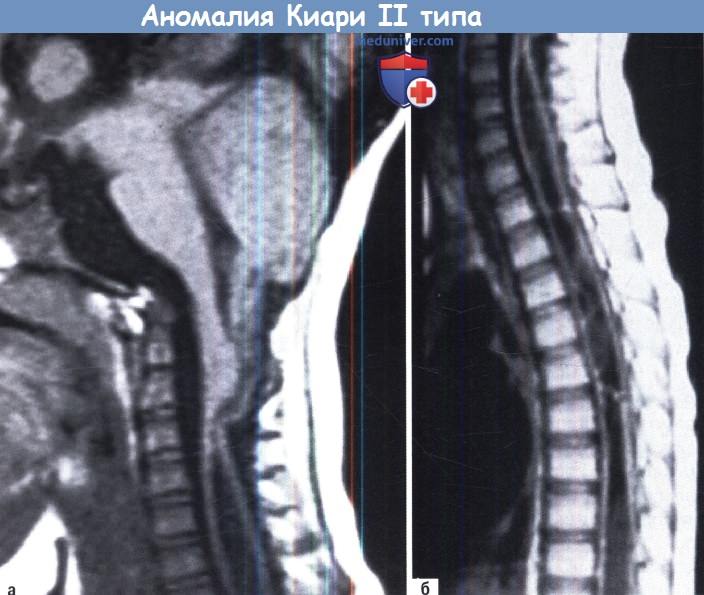
Алгоритм диагностики и лечения аномалии Киари 2 типа — Европейские рекомендацииИзменения в заднем мозге, которые характеризуют аномалию Киари 2 типа, включают более сильное удлинение и каудальное смещение структур задней черепной ямки в шейный канал. Помимо этого наблюдаются как множественные аномалии ствола, мозжечка и большого мозга, так и изменения свода и основания черепа. Основными признаками аномалии задней черепной ямки являются опущение миндалин мозжечка, нижнего червя и четвертого желудочка с его хороидальным сплетением в верхний шейный канал; шейно-мозговая деформация, смещение верхнего червя в увеличенную вырезку намета; перегиб или разветвления сильвиева водопровода; изменения четверохолмия (вторичные по отношению к частичному или полному слиянию холмов). Что касается супратенториальных отклонений, то основные изменения включают гидроцефалию, полигирию, гетеротопию, агенезию мозолистого тела различной выраженности (от частичной до полной), значительное увеличение объема промежуточного вещества. Гидроцефалия присутствует более чем в 90% случаев, она часто асимметричная, с выпуклыми затылочными рогами (кольпоцефалия) и маленьким третьим желудочком. Наконец, костные изменения включают маленькую заднюю черепную ямку, неровную каменистую кость, укороченный скат, расширенное большое затылочное отверстие и краниолакунию. Кроме того, присутствуют изменения ТМО, а именно тенториальная гипоплазия и низкое прикрепление намета (с клочком, в большинстве случаев, лежащим чуть выше или на уровне БЗО), а также гипоплазия серпа большого мозга и его фенестрация. Кроме черепных изменений отличительной чертой в 40-95% случаев является наличие полости в спинном мозге, в основном, на шейном уровне. Аномалия Киари 2 типа почти исключительно всегда связана с менингомиелоцеле. а) Клиническая картина аномалии Киари 2 типа. Клиническая картина варьирует в зависимости от возраста, когда начались проявления. При развитии в раннем детстве в клинической картине преобладают признаки сдавления ствола головного мозга. К ним относятся стридорозное дыхание в результате паралича голосовых связок, центральное обструктивное апноэ; нарушение глотания; нарушения дыхания с возможной потерей сознания, гипотония, тетрапарез и опистотонус. В некоторых случаях, почти исключительно у младенцев и маленьких детей, дисфункция заднего мозга может проявиться остро, сформировав угрожающий жизни синдром, характеризующийся стридорозным дыханием, одышкой, нарушением глотания с аспирационной пневмонией, и опистотонусом, который может прогрессировать вплоть до смерти ребенка, независимо от каких-либо терапевтических процедур. Было показано, что это потенциально катастрофический синдром не соотносится ни с повышением ВЧД, ни с локализацией и степенью спинальных нарушений. У детей старшего возраста или подростков, а также у взрослых лиц молодого возраста в клинической картине, как правило, преобладают спинальные, мозжечковые и офтальмологические симптомы. К ним относятся затылочные и шейные боли, миелопатия со слабостью верхних конечностей, атаксия, косоглазие, нистагм, нарушения слежения, оптокинетических движений и конвергенции, и сколиоз. б) Лучевая диагностика. Основную роль в диагностике аномалии Киари 2 типа играет МРТ. В самом деле, различные супратенториальные и спинальные изменения, а также изменения задней черепной ямки достаточно хорошо визуализируются при этом исследовании. КТ играет важную роль в диагностике костных аномалий основания и свода черепа. в) Лечение аномалии Киари 2 типа. Большинство пациентов имеет гидроцефалию, поэтому, постановка вентрикулярного шунта или оценка функции установленного шунта должны рассматриваться в первую очередь, до декомпрессии задней черепной ямки. На самом деле, симптомы и признаки, связанные с аномалией Киари 2 типа можно просто соотнести с повышением ВЧД вследствие неадекватно функционирующего шунта. Перед проведением хирургической декомпрессии задней черепной ямки необходимо точное планирование операций, так как анатомия этой области значительно изменена (низко лежащий клочок, шейно-мозговая извитость, четвертый желудочек часто находится на шейном уровне, а также наличие аномальных дуральных синусов). Плотные арахноидальные спайки с поверхностной гиперваскуляризацией делают рассечение паутинной оболочки очень трудной и опасной, и процедура должна быть закончена, если обнаружено дно четвертого желудочка. Эффект хирургической декомпрессии зависит от состояния пациента до оперативного вмешательства. Если младенцы, клинические проявления у которых ограничены стридорозным дыханием, имеют все шансы на полное выздоровление, то дети со стридором и приступами апноэ выживают в 75% случаев, и только у 50% из них наступает полное выздоровление. Младенцы со стридором, приступами апноэ, дисфагией и синюшностью имеют только 40% шанс на выживание при минимальном шансе функционального восстановления. Такой неблагоприятный исход можно объяснить либо обширным и необратимым повреждением структур ствола мозга, либо анатомическими нарушениями этих структур, не поддающимися какой-либо хирургической коррекции.
— Также рекомендуем «Алгоритм диагностики и лечения аномалии Киари 3 типа — Европейские рекомендации» Оглавление темы «Аномалия Киари.»:
|
Источник

Аномалия Киари (синдром Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
Общие сведения
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.

Аномалия Киари
Причины возникновения аномалии Киари
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация аномалии Киари
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Диагностика аномалии Киари
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз аномалии Киари
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Источник
 Аномалия Арнольда-Киари – это нарушение функций мозга. Оно проявляется с самого рождения и идентифицируется отличием от нормальных параметров затылочной черепной ямки и составляющих мозга, которые расположены в этом районе.
Аномалия Арнольда-Киари – это нарушение функций мозга. Оно проявляется с самого рождения и идентифицируется отличием от нормальных параметров затылочной черепной ямки и составляющих мозга, которые расположены в этом районе.
Такие нарушения ведут к тому, что миндалины мозжечка опускаются в затылочную часть и ущемляются там.
В этом случае они спускаются таким образом, что достигают уровня первого или второго шейных позвонков и блокируют нормальное протекание спинно-мозговой жидкости.
На данный момент причины возникновения этого заболевания полностью не изучены, хотя сейчас их выделяют несколько разновидностей.
Аномалия Арнольда Киари 1 степени – это врождённые изменения, которые передались от родителей. Аномалия Арнольда Киари 2 степени – это травмы, которые получил ребёнок при рождении.
Статистика говорит, что от этого заболевания страдают в 3,3-8,2 случаев из 100000 наблюдавшихся. В среднем чаще от этого заболевания страдают люди 25-40 лет.
Типы болезни и особенности каждого из них
В 1891 году учёным Киари были выделены четыре вида данного заболевания. Эта классификация актуальна и в наши дни, поэтому врачи ею активно пользуются:
- Аномалия Арнольда Киари 1 типа— для него характерно снижение вдоль оси позвоночного столба составляющих затылочной черепной ямки до уровня первого или второго позвонка.
- II тип – очень часто можно встретить развитие гидроцефалии.
- III тип – встречается не так часто, как первые два, и характеризуется значительным перемещением большинства составляющих задней черепной ямки.
- IV тип – происходит гипоплазия мозжечка, при этом он не смещается вниз.
Если у человека наблюдается аномалия III или IV типа, то жить с ней невозможно.
Причины возникновения болезни
Нельзя наверняка сказать из-за чего может развиться аномалия Арнольда-Киари. Может быть, что вероятность рождения ребёнка с этим заболеванием увеличивают такие факторы, которые имеют влияние на организм женщины, которая ждёт ребёнка:
- употребления лекарственных препаратов выше допустимой нормы;

- размеры задней черепной ямки слишком малы;
- приём алкогольных напитков и курение в период беременности;
- частое поражение организма вирусными простудными заболеваниями;
- возникновение повреждений черепного отдела или головного мозга как результат родовой травмы;
- бывают врождённые и приобретённые причины появления аномалии Арнольда-Киари.
Симптомы и признаки
Чаще всего можно встретить аномалии Арнольда-Киари I типа. Зачастую она даёт о себе знать в период полового созревания или уже у взрослых. В таком случае проявляются следующие симптомы аномалии Арнольда-Киари:
- головные боли, возникающие в результате кашля, чихания или физических нагрузок;
- нарушение нормальной походки, вызванное проблемами с поддержанием равновесного состояния;
- проблемы с мелкой моторикой рук;
- нарушенная температурная чувствительность;
- онемение в области кистей рук;
- ощущение болей в области затылка и шеи.
Что касается II и III типов этого заболевания, то они проявляются такими же симптомами, только это происходит ещё с самого рождения.
В чём состоит сложность диагностики?
Очень сложно диагностировать аномалию Арнольда-Киари. Если делать неврологическое обследование, то оно сможет указать только на повышение уровня внутричерепного давления, которое называется гидроцефалией.
Если делать рентген головного мозга, то он диагностирует только нарушение строения костей, которыми обычно сопровождается это заболевание.
Единственный метод, который даёт возможность точно определить аномалию Арнольда-Киари – это магнитно-резонансная томмография. Для того, чтобы получить правильный и достоверный результат, нужно чтобы пациент лежал неподвижно. Для этого дети должны находится в медикаментозном сне.
Иногда это заболевание может себя не проявлять абсолютно никаким образом. И тогда его можно диагностировать только в результате усиленного медицинского осмотра.

На фото МРТ при аномалии Арнольда Киари
Лечение болезни
Существует два способа лечения аномалии Арнольда-Киари – консервативный или нехирургический и хирургический.
В случае, когда это заболевание протекает без проявления каких-либо симптомов, тогда оно не нуждается в лечении.
Если аномалия Арнольда-Киари проявляется только болевыми ощущениями в затылочной области и шее, то в этом случае назначается консервативное лечение. Тогда назначаются обезболивающие или противовоспалительные препараты. Это и есть нехирургический способ лечения.
В случае, когда симптомы болезни не поддаются консервативному лечению, тогда назначают хирургическое.
Целью такого лечения является устранение факторов, из-за которых происходит сжимание структур головного мозга.
В результате проведения операции расширяется затылочная часть головы, улучшается передвижение цереброспинальной жидкости по организму. Возможно проведение шунтирующих операций.
Осторожно, видео операции! Кликните, чтобы открыть
Осложнения, которые может вызвать заболевание
В некоторых случаях аномалия Арнольда-Киари может быть очень прогрессивно развивающимся заболеванием и привести к определенным осложнениям. Ими могут быть:
- Гидроцефалия, возникающая в результате накопления большего чем допускается количества жидкости.
- Паралич, причиной которого может выступать сдавливание спинного мозга.
- Сирингомиелия, что подразумевает образование полости или кисты в позвоночнике. В такие полости, после их возникновения, может поступать жидкость и приводить к нарушению нормальной работы спинного мозга.
- Возможно нарушение дыхательного процесса, которое развивается вплоть до его остановки.
- Могут быть случаи застойной пневмонии, возникающие как результат того, что пациент теряет способность передвигаться самостоятельно.
Прогноз
Бывают случаи, когда аномалия Арнольда-Киари I типа не проявляет свои симптомы на протяжении всей жизни человека, имеющего это заболевание.
Аномалия Арнольда-Киари III типа чаще всего приводит к неблагоприятному исходу.
Если при аномалии Арнольда-Киари I и III типов возникают какие-либо новрологические симптомы, то необходимо как можно раньше провести хирургическое лечение.
Это нужно потому, что такой неврологический недостаток очень сложно полностью устранить даже после успешного проведения операция, особенно если она была несвоевременной или затянутой.
Статистика показывает, что эффективность проведения хирургического лечения составляет 50-85%.
Источник