Синдром апера код мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Апера.
Синдром Апера
Описание
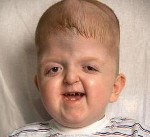
Синдром Апера. Генетическое заболевание, характеризующееся нарушениями процессов окостенения черепа и связанными с этим вторичными расстройствами, а также многочисленными пороками развития скелета и конечностей. Симптомами этого состояния являются карликовый рост, башенная форма черепа, расширенная переносица, незаращение твердого нёба, синдактилии на руках и ногах. Диагностика синдрома Апера производится по характерной клинической картине патологии, на основании рентгенологических данных и молекулярно-генетических исследований. Специфического лечения заболевания не существует, применяют поддерживающую терапию, проводят хирургические вмешательства паллиативного характера.
Дополнительные факты
Синдром Апера (акроцефалосиндактилия 1 типа) – генетическая патология, обусловленная нарушением образования некоторых видов соединительной ткани, главным образом костной. Впервые данное состояние было описано в 1906 году французским педиатром Э. Апером, дальнейшие исследования подтвердили генетическую природу этого заболевания. Этиология и молекулярно-генетические механизмы развития синдрома Апера были определены значительно позднее – лишь в 1995 году. Данная патология может наследоваться по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев ее причиной являются спонтанные мутации в половых клетках родителей (так называемые герминативные мутации).
Синдром Апера с одинаковой частотой поражает как мальчиков, так и девочек, его встречаемость составляет в среднем 1 случай на 160 000-200 000 новорожденных. Врачи-генетики в настоящее время относят синдром Апера к особой группе наследственных заболеваний – акроцефалосиндактилиям, характеризующиеся одновременным поражением костей черепа и конечностей. Особенностью этой патологии является важность ее как ранней диагностики, поскольку паллиативные мероприятия в раннем возрасте могут в значительной степени влиять на дальнейшее интеллектуальное развитие больного.
Синдром Апера
Причины
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной. Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями. Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R. Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния. Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности – к преждевременному зарастанию швов и остановке нормального роста черепной коробки. Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов. Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR. Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей – различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Симптомы
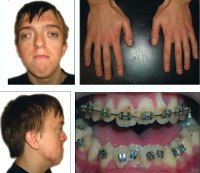
Некоторые проявления синдрома Апера заметны с самого рождения – например, синдактилия, которая может быть полной или в виде перепонок. Как правило, срастаются 2, 3 и 4 пальцы на кистях, иногда аналогичный порок возникает и на пальцах ног. Среди неонатологов симптом иногда носит название «среднего пальца» – в тяжелых случаях эти три пальца прочно срастаются между собой и имеют один общий ноготь. Другим постоянным симптомом синдрома Апера, обнаруживающимся сразу после рождения или в первые месяцы жизни, является раннее развитие синостоза костей черепа. Чаще всего происходит срастание венечного или стреловидного шва, что по мере роста головного мозга приводит к деформации черепа по типу «башенной». Из-за черепного синостоза у больных синдромом Апера наблюдается хроническое повышение внутричерепного давления, становящееся причиной задержки умственного развития, головных болей, тошноты и рвоты.
Помимо деформации черепа о наличии синдрома Апера свидетельствует характерный внешний вид больных. У них обычно обнаруживается плоский или выпуклый лоб, гипертелоризм и экзофтальм, может развиваться косоглазие. Деформации затрагивают и кости лицевого черепа – переносица расширена, челюсти нередко недоразвиты, наблюдается нарушение прикуса. Из других симптомов синдрома Апера иногда регистрируются нарушения дыхания (из-за недоразвития верхней челюсти, сужения хоан или трахеи), незаращение твердого нёба, врожденные пороки сердца, аномалии развития позвонков, почек, прямой кишки.
Рвота. Тошнота.
Диагностика
Диагностика синдрома Апера производится на основании осмотра и изучения настоящего статуса пациента, рентгенологических исследований, молекулярно-генетических анализов. При осмотре у больного выявляется синдактилия (у лиц старшего возраста могут обнаруживаться следы ее хирургической коррекции), деформация черепа – башенный череп или брахикефалия, характерный внешний вид лица. С возрастом у больных синдромом Апера могут нарастать признаки нарушения дыхания, при ЭхоКГ нередко определяются пороки сердца и сосудов – стеноз легочного ствола или аорты, дефекты межжелудочковой перегородки. Иногда на этом фоне выявляются признаки сердечной недостаточности. Также возможно наличие иных пороков развития – аномалий позвонков, глухоты, слепоты (из-за катаракты, пигментного ретинита, атрофии зрительных нервов), патологий почек и поджелудочной железы. Из-за столь широкого спектра возможных нарушений больные синдромом Апера нуждаются в тщательном и всестороннем медицинском обследовании.
Рентгенологическими методиками уже у маленьких детей можно обнаружить синостоз костей черепа в области венечного или стреловидного шва. В дальнейшем при помощи рентгенографии можно определить характерную для синдрома Апера деформацию черепной коробки, пороки развития костей лицевого черепа, аномалии позвонков и другие нарушения. Наиболее достоверным диагностическим методом при этом состоянии является молекулярно-генетический анализ. Как правило, для выявления синдрома Апера производят секвенирование 7 экзона гена FGFR2, иногда используют менее затратные техники, ориентированные только на поиск наиболее распространенных мутаций (S252W и P253R), приводящих к этому заболеванию. Подобные методики более дешевые и быстрые в выполнении, обладают точностью на уровне 95%, возможно их использование в качестве пренатальной диагностики этого состояния. Подобный анализ особенно актуален, если посредством профилактических УЗИ у плода выявляются нарушения, предположительно связанные с синдромом Апера – пороки развития черепа, сердца, верхних или нижних конечностей.
Лечение
Специфического лечения синдрома Апера на сегодняшний день не существует, однако паллиативные и симптоматические мероприятия могут значительно облегчить состояние больного и улучшить качество его жизни. Особенно важно как можно раньше диагностировать это заболевание по той причине, что своевременная хирургическая коррекция черепного синостоза позволит избежать значительного роста внутричерепного давления. По многочисленным данным, после таких операций, произведенных в раннем детстве, признаки умственной неполноценности у больных синдромом Апера были выражены значительно слабее, иногда сохранялся нормальный интеллект. Поэтому борьба с внутричерепной гипертензией играет центральную роль в паллиативном лечении этого состояния. Если же у пациентов имеется умственная отсталость, то ее выраженность снижается путем психокоррекционной работы.
Другой часто выполняемой паллиативной хирургической операцией при синдроме Апера является вмешательство для разделения сросшихся пальцев на руках и ногах. Это относительно несложная процедура при перепончатом типе сращения, однако при более тяжелых формах порока операция значительно усложняется. При синдроме Апера также может потребоваться помощь хирургов в случае пороков сердца, сужения хоан или трахеи, нарушения формирования прямой кишки и других проявлений этого генетического заболевания. Больные нуждаются в регулярных медицинских обследованиях у специалистов различного профиля.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа. Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком. Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Апера (Аперта) является генетической патологией, в результате которой наблюдаются пороки в развитии черепа (широкопосаженные глаза, излишне высокий череп), а помимо этого нижних и верхних конечностей (пальцы на них полностью срастаются; могут появляться также дополнительные).
 [1], [2], [3]
[1], [2], [3]
Код по МКБ-10
Q87 Другие уточненные синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Эпидемиология
Эпидемиологические показатели: подобные случаи встречаются 1 на 160 тыс. Больные с данной патологией составляют примерно 4,5% ото всех случаев развития краниосиностозов.
[4], [5], [6], [7], [8], [9]
Причины синдрома Апера
Развитие синдрома провоцируется нарушением в строении гена, находящегося в 10-й хромосоме. Данный ген ответственен за процесс разделения пальцев, а помимо этого за своевременное закрытие черепных швов.
Помимо этого причинами появления патологии считаются перенесённые в период вынашивания инфекционные болезни (такие, как краснуха или туберкулёз, сифилис либо грипп, а также менингит), облучение матери рентгеновскими лучами. Чаще всего подобный синдром выявляют у детей, родившихся у пожилых родителей.
[10], [11], [12], [13], [14], [15]
Патогенез
Синдром Апера передаётся наследственным путём. Его тип – аутосомно-доминантный (т.е., в семье, в которой болен кто-то из будущих родителей, вероятность рождения малыша с этим заболеванием равна 50-100%).
Уникальный фактор роста фибробластов рецептор 2 (FGFR2) вследствие мутации приводит к увеличению числа клеток-предшественников, которые развиваются по остеогенному пути. В конечном счете, это приводит к повышенному образованию поднадкостничной костной матрицы и преждевременному окостенению швов черепа во время развития плода. Порядок и скорость шовного плавления определяют степень деформации и инвалидности. После того, как шовный материал заростает, рост других тканей перпендикулярно к этому шву становится ограниченным, и слитые кости действуют как единый костный каркас.
Первое генетическое доказательство того, что синдактилия при синдроме Апера является следствием дефекта фактора роста рецептора кератиноцитов (KGFR) стало наблюдение корреляции между экспрессией KGFR в фибробластах и серьезности синдактилией.
Амблиопия и косоглазие чаще встречается у пациентов с мутацией FGFR2 Ser252Trp а атрофия диска зрительного нерва чаще встречается у пациентов с мутацией FGFR2 Pro253Arg. У больных с FGR2 Ser252Trp мутации имеют значительно большую распространенность нарушений зрения по сравнению с пациентами с мутацией FGFR2 Pro253Arg.
[16], [17], [18], [19], [20], [21], [22]
Симптомы синдрома Апера
Отдельные проявления болезни отчётливо заметны уже при рождении малыша, потому как развиваются ещё внутри материнской утробы. Среди основных симптомов синдрома:
- Череп деформируется – вытягивается в высоту, приобретая вид «башенного»; помимо этого глаза широко посажены и слегка выпучены (потому как размер глазных орбит уменьшается); расширяется нос и формируется неправильный прикус (верхние зубы чересчур выпирают);
- Пальцы конечностей полностью срастаются (в основном это безымянный, а также средний вместе с указательным) и выглядят как кожная перепонка либо полное сращение костей; помимо этого могут вырастать лишние пальцы;
- Задержка в умственном развитии (происходит не у всех);
- Зрительные нервы атрофируются, вследствие чего острота зрения снижается (в отдельных случаях развивается полное отсутствие зрения);
- Увеличение внутричерепного давления, происходящее в результате слишком раннего зарастания черепных швов – проявляется в виде головных болей и рвоты с тошнотой;
- Так как верхняя челюсть осталась недоразвитой, появляются проблемы с дыханием;
- Синдром апноэ сна является распространенным явлением.
- Эмоциональные проявления – агрессия, отсутствие сдержанности, сильная вспыльчивость.
[23], [24], [25], [26], [27], [28]
Осложнения и последствия
Большинство пациентов имеют некоторую степень обструкции верхних дыхательных путей в грудном возрасте. Верхние дыхательные пути за счет уменьшения размера носоглотки и хоан имеют плохую проходимость, нижние дыхательные пути в связи с аномалиями трахеи хряща могут стать причиной ранней смерти.
[29], [30], [31], [32], [33], [34], [35]
Диагностика синдрома Апера
Для постановки диагноза требуются следующие мероприятия:
- Врач должен проанализировать жалобы пациента, а также анамнез болезни. Необходимо выяснить, имелись ли в семье случаи развития подобной патологии;
- Неврологическое обследование, чтобы оценить форму черепа и интеллектуальное развитие пациента (специальные анкеты, а также беседа);
- Обследование глазного дна, чтобы выявить наличие симптомов увеличения внутричерепного давления (отёк ДЗН, а также размытость его краёв);
- Для оценки состояния черепа выполняется его рентгенография;
- Компьютерная, а также магнитно-резонансная томография головы, чтобы послойно обследовать структуру головного мозга с черепом, определить наличие симптомов преждевременного сращения черепных швов, а помимо этого гидроцефалию (из-за увеличения внутричерепного давления скапливается излишек ликвора (это цереброспинальная жидкость, которая способствует процессу обмена веществ, а также питанию головного мозга));
- Рентген стоп с кистями, чтобы узнать причину, из-за которой произошло сращение пальцев (это важно для планирования последующего хирургического вмешательства);
- Может быть назначена консультация у медицинского генетика, а также нейрохирурга.
[36], [37], [38], [39], [40], [41]
Анализы
Проводится генетический анализ частых мутаций, возникающих в гене типа FGFR2.
[42], [43], [44], [45], [46], [47], [48]
Дифференциальная диагностика
Дифференцировать этот синдром нужно с прочими генетическими патологиями, при которых наблюдается краниосиностоз. Это такие болезни, как синдромы Пфайффера, Крузона, а также Сэтре-Чотцена и Карпентера. Чтобы исключить данные аномалии, применяются молекулярно-генетические способы тестирования.
Лечение синдрома Апера
Выполнение хирургической операции считается единственным действенным методом лечения синдрома Апера – он помогает откорректировать отдельные физические дефекты, а также исправляет отсталость в умственном развитии.
В процессе проведения данной процедуры выполняется закрытие венечного шва, чтобы предупредить возможное травмирование головного мозга. Наиболее распространённой считается методика краниофациальной дистракции, при которой выполняется градационное вытяжение черепа. Для удаления отдельных дефектов на лице проводится ортодонтическая и/либо ортогнатическая операция.
Помимо этого пациентам хирургическим путём устраняют сращение пальцев.
Профилактика
Так как синдром Апера является наследственной патологией, способов профилактики его появления не существует.
[49], [50], [51], [52], [53], [54], [55], [56]
Прогноз
Синдром Апера имеет неблагоприятный прогноз дальнейшего развития и жизни ребёнка. Исключение составляют лишь дети, которые достигли зрелости без каких-либо сердечных патологий.
[57], [58], [59], [60], [61], [62]
Источник