Семячкина а н и синдром марфана

Болезнь Марфана, или синдром Марфана, относится к числу наследственных болезней соединительной ткани. Клиническое описание этого заболевания было дано впервые фран-цузским педиатром А. Марфаном (1896). Ашар (1902) назвал его арахнодактилией и долихосте-номелией. Второе наиболее известное название — синдром Марфана — предложено Бергером (1915).
В этиологии и патогенезе синдрома Марфана большое значение придают поражению со-единительной ткани, а именно нарушению синтеза коллагена, которое приводит к накоплению фракций растворимого (незрелого) коллагена с распадом его на метаболиты, содержащие окси-пролин.
Заболевание проявляется уже в детском возрасте, но большинство пациентов — молодые люди. Нередко болезнь приводит к ранней инвалидизации и ранней смерти; встречается с рав-ной частотой у мужчин и женщин.
Типичным для синдрома Марфана считают сочетание характерных изменений опорно-двигательного аппарата (долихостеномелия, арахнодактилия), глаз (подвывих хрусталика) и сердечнососудистой системы (эрдхеймовский некроз, аневризма аорты).
Доминантный характер наследования предполагает как один из механизмов естественно-го отбора наличие стертых форм заболевания, диагностика которых достаточно сложна. Многие биохимические критерии диагностики, предлагаемые для синдрома Марфана, неспецифич-ны для него, а определение оксипролина настолько трудоемко, что не может быть использова-но в исследованиях. Около 40 % больных имеют стертую форму заболевания, при которой отсутствуют выраженные изменения в какой-либо системе органов.
Большая часть работ в литературе на эту тему посвящена детям с синдромом Марфана, так как ранняя диагностика необходима для разграничения с фенотипически схожим наследст-венным заболеванием обмена метионина — гомоцистинурией, а также для своевременного меди-ко-генетического консультирования и прогнозирования будущего потомства.
Поздняя диагностика синдрома Марфана (у взрослых лиц) может свидетельствовать лишь о неудовлетворительном функционировании медико-генетической службы. Классический синдром Марфана не представляет больших затруднений для диагностики.
Целью работы было обобщение данных литературы и результатов собственных наблюдений болезни Марфана, выделение стертых форм ее, определение критериев диагностики, приведение рабочей классификации заболевания и решение вопросов динамического наблюдения.
При наличии классической триады симптомов и семейного характера заболевания диаг-ноз в большинстве случаев не вызывает сомнений, но, учитывая разноплановость этих измене-ний, одному специалисту не всегда удается заподозрить данное заболевание.
Клинические признаки синдрома Марфана


Примечание. + наличие, — отсутствие данного признака.
Под нашим наблюдением находились б мужчин в возрасте от 18 до 20 лет. Поводом для госпитализации послужили усиление кардиалгий, одышки, появление быстрой утомляемости при физических нагрузках. 3 человека поступили в отделение торакальной хирургии с диагнозом: воронкообразная грудная клетка с нарушением функции дыхания; 2 — в кардиологическое отде-ление с нейроциркуляторной дистонией по кардиальному типу; 1 больной острым бронхитом — в пульмонологическое отделение. Исходя из медицинских документов, все они считались здоро-выми. Таким образом, на догоспитальном этапе заболевание соединительной ткани — синдром Марфана — распознано не было. Диагноз верифицирован с помощью клинических и инструмен-тальных (рентгенография опорно-двигательного аппарата, ультразвуковое исследование сердца) методов с привлечением специалистов (отоларинголога, окулиста, невропатолога, нейро-хирурга, травматолога).
Данные о клинических и инструментальных признаках каждого больного с синдромом Марфана представлены в таблице. Учитывая эти данные, а также результаты исследований дру-гих авторов, критериями отбора лиц с возможным синдромом Марфана следует считать:
1. Семейный анамнез (наличие родственников с синдромом Марфана).
2. Изменение опорно-двигательного аппарата (высокий рост, чаще выше 190 см), несо-ответствие между ростом и массой тела в сторону ее уменьшения, длинные тонкие конечности, особенно с выраженным удлинением дистальных отделов, узкие кисти с длинными пальцами (арахнодактилия), плоскостопие, долихоцефалия, грыжи, идиопатические сколиозы, остеохонд-роз позвоночника, часто с грыжами Шморля, деформации грудной клетки, совокупность пере-численных изменений.
3. Нарушения со стороны глаз (подвывих хрусталика, высокая степень миопии, миопи-ческий астигматизм).
4. Изменения со стороны сердечно-сосудистой системы. Клинические: пролабирование митрального и других клапанов, недостаточность аортального клапана, быстро прогрессирую-щие кардиалгии неуточненной этиологии, особенно у лиц молодого возраста, аневризма аорты, особенно у молодых людей. Выявляемые специальными методами исследования: эхокардиография: пролабирование митрального клапана, других клапанов, нескольких клапанов; расширение кольца аорты и диаметра аорты, ее усиленная пульсация, признаки регургитации. Патолого-анатомически: аневризмы разных отделов аорты, разрыв аорты без анамнеза аортальной гипер-тензии у лиц без признаков атеросклероза аорты. Гистологически: эрдхеймовский некроз.
5. Изменения со стороны органов дыхания: спонтанный пневмоторакс, поликистоз лег-ких, ранние идиопатические формы эмфиземы, буллезная эмфизема. Рентгенологически: широкие межреберные промежутки, деформированные ребра, грудина, сколиоз.
6. Нарушения со стороны желудочно-кишечного тракта: висцероптоз, большое количест-во мукоидных веществ в желудочном содержимом.
7. Изменения ЛОР-органов и полости рта: готическое небо у лиц с узкими челюстями, неправильный прикус, уменьшение толщины костей черепа.
8. Рентгенологические находки: тонкие длинные трубчатые кости с поперечной исчер-чен-ностью и остеопорозом метафизов, изменение размеров передней черепной ямки, удлинен-ный канал спинного мозга, длинные тонкие кости кистей, стоп.
В отношении критериев диагноза существуют определенные разногласия между исследо-вателями. Ранее достоверными считались лишь случаи с выраженными проявлениями синдрома — сочетанием изменений в трех системах, которые у ряда пациентов могут наблюдаться уже в детском возрасте.
Однако большинство авторов, учитывая генетическую природу заболевания и особенно-сти характеристики доминантного гена, признают наличие неполных форм заболевания ив спорадических случаях. Биохимические критерии (изменения содержания глюкозаминогли-канов, оксипролина) важны, но, к сожалению, трудоемкость соответствующих анализов и не-возможность их осуществления в большинстве лечебных учреждений снижают их практическую ценность.
Доступными тестами для определения арахнодактилии являются:
— тест большого пальца, предложенный в 1945 г. (Parker, Haze, 1945): разогнутый первый палец приводится к кисти. Если тест положительный, палец выступает за мягкие ткани кисти. В сомнительных случаях необходима рентгенография кисти с приведенным большим паль-цем — фаланга большого пальца при положительном тесте выступает за скелет метакарпальных костей;
— тест запястья: при обхватывании пальцами растянутой кисти одной руки»запястья другой при арахнодактилии первый и пятый пальцы легко соединяются друг с другом.
Другие тесты, которые можно использовать для диагностики синдрома Марфана:
— соотношение кисть — рост более 11 %;
— соотношение ступня — рост более 15 %;
— размах рук больше роста на 5 см;
— длина среднего пальца больше 10 см;
— расстояние от лобка до пола превышает ‘/г роста более чем на 6 см.
Рентгенологический тест: метакарпальный индекс больше 8,4.
Если размах рук превышает рост более чем на 5 см, длина ног от лобка до пола более ‘/г роста, метакарпальный индекс больше 8,4, а также положителен результат теста большого пальца, диагноз синдрома Марфана следует считать достоверным.
Достоверность диагноза увеличивается при обследовании семьи.
Часто среди молодых людей выявляют кифосколиозы, некоторую гипермобильность суста-вов, плоскостопие, повышенную растяжимость связок, высокое небо, голубые склеры в сочета-нии с миопией, пролапс митрального клапана, мягкие ушные раковины. Комбинация этих при-знаков различна, так же как и степень их выраженности. О. В. Лисиченко относит таких больных к марфаноподобному варианту.
Таким образом, следует выделять следующие клинические варианты болезни:
1. Болезнь Марфана (наличие классических трех признаков, семейный характер заболе-вания).
2. Синдром Марфана (наличие стертых форм с положительными вышеперечисленными диаг-ностическими тестами).
3. Марфаноподобный синдром. Классификация болезни (синдрома) Марфана
(О. В. Лисиченко, 1986).
I. Форма
Стертая: слабо выраженные изменения в одной, двух системах. Выраженная:
1. Слабо выраженные изменения в трех системах.
2. Выраженные изменения хотя бы в одной системе (ограниченная форма).
3. Выраженные изменения в двух, трех системах и более.
Степень выраженности: а) легкая; б) средняя; в) тяжелая.
II. Характер течения
Рецидивирующий (прогрессирующий). Стабильный
III. Генетическая характеристика
Семейная форма (тип наследования). Первичная мутация.
Примерные диагнозы:
— болезнь Марфана, выраженная форма с преимущественным поражением сердечно-сосудистой системы средней степени тяжести, прогрессирующее течение, семейный случай, аутосомно-доминантный тип наследования:
— синдром Марфана, выраженная форма с преимущественным поражением глаз (подвывих хрусталика, миопатический астигматизм), сердечнососудистой системы (пролабирование мит-рального клапана с регургитацией (-|—(-), Hi), опорно-двигательного аппарата (долихосте-номе-лия, остеохондроз грудного отдела позвоночника без нарушения функции), дыхательной системы (диффузная эмфизема с нарушением функции внешнего дыхания I степени), спорадиче-ская форма;
— марфаноподобный синдром с преимущественным поражением опорно-двигательного аппа-рата (долихостеномелия, остехондроз грудного отдела позвоночника без нарушения функции), дыхательной системы (диффузная эмфизема легких с нарушением функции внешнего дыхания I степени), спорадическая форма;
— марфаноподобный синдром с преимущественным поражением опорно-двигательного аппа-рата (кифосколиоз, плоскостопие II степени), глаз (миопия высокой степени), сердечно-сосудистой системы (пролабирование митрального клапана без регургитации).
Вероятность правильного диагноза повышается при наличии совокупности признаков. В связи с этим необходимо оценивать значимость каждого признака с выяснением семейного анамнеза.
Диагноз выраженной формы при легкой степени изменений 3—4 систем и тем более диаг-ноз стертых форм правомочны в настоящее время, по мнению исследователей, только для род-ственников больных.
Со стороны сердечно-сосудистой системы к легким изменениям можно отнести начальную стадию пролабирования митрального клапана, к выраженным — пролабирование митрального и трех-куспидального клапанов, расширение аортального кольца, к тяжелым — аневризму аорты.
К легкой степени изменений опорно-двигательного аппарата относятся нарушения про-порций, воронкообразная деформация грудной клетки и сколиоз I степени; к средней степени — воронкообразная деформация, остеохондроз позвоночника с грыжами Шморля, сколиоз II сте-пени, плоскостопие, грыжи; к тяжелой — кифосколиоз III—IV степени, воронкообразная дефор-мация III—IV степени.
Со стороны глаз к изменениям легкой степени можно отнести гетерохромию радужки, малую степень миопии; к выраженным — подвывих хрусталика, миопический астигматизм, миопию высокой степени; к тяжелым — наличие осложнений в виде вывиха хрусталика, отслойки сет-чатки.
На основании собственных наблюдений и обобщенных данных литературы нами выделены и систематизированы (см. таблицу) наиболее частые признаки стертых форм синдрома Марфана, совокупность которых поможет упростить клинический, вариант дифференциальной диагностики.
При подозрении на синдром Марфана целесообразно использовать перечисленные в таб-лице изменения в комплексе. Критерии диагноза для родственников могут быть менее жестки-ми, когда родословная становится дополнительным, а иногда и решающим тестом для уточнения диагноза.
Лица с подозрением на синдром Марфана подлежат диспансерному динамическому наблю-дению с осмотром терапевтом через каждые 6 мес и другими специалистами (по показаниям), а также последующему (после постановки диагноза) медико-генетическому консультированию для лиц репродуктивного возраста.
Правильная диагностика и интерпретация клинических данных позволяют в ряде случаев уточнить диагноз синдрома Марфана, оценить функциональное состояние органов и систем, определить риск возможных осложнений, а поэтому играют существенную роль в распознавании заболевания в молодом возрасте и разработке профилактических мероприятий.
ЛИТЕРАТУРА
1. Бочкова Д. #., Артамонова Н. П., Кузьмина П. И. и др. Пролапс митрального клапана как симптом наследственных заболеваний // Вопр. охр. мат.— 1979.— № 10.— С. 12—18.
2. Гарпузов В. В. Клинико-биохимическое изучение синдрома Марфана: Автореф. дис. … канд.— Л., 1973.
3. Гордое И. Б., Ляхер А. В. Поражение сердечно-сосудистой системы при синдроме Марфана // Клин. мед.— 1980,— № 8.— С. 98—100.
4. Гофман В. А., Коробейникова С. А., Могилевский Р. Э. О клинической симптоматике стертых форм синдрома Марфана // Там же.— 1979.— № 6.— С. 90—92.
5. Казначеева В. П., Лисиченко О. В. Клинико-генетиче-ское обследование больных с синдромом Марфана. // Научная конф. по клинической генетике: Материалы.— М., 1971.—С. 9—11.
6. Лисиченко О. В. Синдром Марфана.— Новосибирск: Наука, 1986.
7. Наследственные системные заболевания скелета / Волков М. В., Меерсон Е. М., Негволдова О. Л. и др.— М.: Медицина, 1982.
8. Семячкина А. Н. Принципы диагностики синдрома Марфана: Автореф. дис. … канд.— М., 1975.
Поступила 18.11.91
Источник
Синдром Марфана (СМ) — наследственное заболевание соединительной ткани с поражением скелетной, мышечной, сердечно-сосудистой систем и глаз, наследуется по аутосомно-доминантному типу. Несмотря на то, что СМ был описан более 100 лет назад, он продолжает оставаться одной из актуальных проблем медицины с позиции этиологии, патогенеза, морфогенеза, диагностики и лечения. В связи с этим до сих пор пересматривают диагностические критерии, классификацию СМ, изучаются фенотипические проявления заболевания и ведутся поиски новых методов лечения. У одного пациента с СМ может быть столько проблем со здоровьем, сколько специалистов в поликлинике.
Историческая справка. В
1896 году
французский педиатр Антуан Жан Марфана описал пятилетнюю девочку с чрезвычайно тонкими и длинными конечностями, контрактурами суставов, кифосколиозом и назвал этот синдром pattes d’araignee (пальцы паука). Эта девочка могла иметь арахнодактилию с контрактурами (синдром Билса), но имя Марфана было использовано для обозначения сочетание симптомов, связанных с дефектами в гене FBN1. Позже Ашари [Achard, 1902] назвал его арахнодактилия и долихостеномелия (греч. dolicos — длинные, stenos — тонкие, melis — конечности).
Существуют другие названия
,
которые составляют сегодня лишь исторический интерес:
— синдром Марфана-Эшера;
— акромакрия Пфаундлера;
— акродолихия Бругшмеллера;
— долихостеномелия;
— синдром Марфана-Эрбе;
— гиперхондроплазия Мэри и Бабоне;
— акрохондрогиперплазия Валентина;
— врожденная акромакрия с амиотонией Юнга;
— врожденная мезодермальная дистрофия Веве;
— дисмезектопия Лавал;
— долихоморфия.
Кроме этого, часть авторов считает, что современное определение синдрома Марфана не имеет никакого отношения к описанному Марфаном в 1896 году [Лисиченко О.В., 1986].
Поражение аорты, как симптом СМ, было выявлено только в
1943 году
EtterLE, GloverLP, а роль дилатации аорты в сокращение продолжительности жизни уточнена в
1972 году
MurdochJL. Роль фибриллина в патогенезе СМ описана
1990 году
Холлистер ET, а также был определен локус этого заболевания хромосоме 15q21.1. Доказательство того, что мутации гена фибриллина-1 (FBN1) могут привести к СМ были описаны в 1991 году Даецом [MosheFrydman, 2008].
Синдром Марфана — редкое заболевание. Частота возникновения 2-3 случая на 10 000. Большинство авторов описывали единичные случаи заболевания.Наибольшее количество случаев описано Марфаном — 10 (1938), Грималди — 15 (1964), Маккьюсикои — 75 пробандов (1966). Отечественные исследователи диагностировали синдром Марфана у 17 больных — (Кондрашин, Неудахин, 1968); 11 — (Надарейшвили, Паламарчук, 1970); 19 — (Добро, Добро, 1970); 30 — (Гусева, 1971); 64 — (Лисиченко, 1971), 23 — (Уткин, Грушин, 1972); 10 — (Гапузов, 1973), 70 — (Семячкина, 1975) [ЛисиченкоО.В, 1986, Барашнев Ю.И. и др., 1983].
Генетика. Генетический характер СМ
впервые заметил Weve H.
, который описал семью с несколькими больными и таким образом доказал аутосомно-доминантный тип наследования [Лисиченко О.В., 1986]. Дальнейшие генетические исследования показали, что мутация в гене фибриллина-1 (FBN1) оказывается не только у больных СМ, но при других похожих заболеваниях соединительной ткани, объединенные в группу фибрилинопатий I типа [Фищеко Я. В., 2006]. Ген FBN1 размещается на длинном плече 15 хромосомы и картирована в локусе 15q 21.1 (рис. 1).
Рисунок 1. Структура пятнадцатой
хромосомы человека
 Примечание. 15-я хромосома человека — одна из 23 человеческих хромосом. Она содержит более 102 млн. пар оснований, что составляет 3-3,5% всего материала ДНК клетки человека. Вероятно, она состоит из 700-900 генов.
Примечание. 15-я хромосома человека — одна из 23 человеческих хромосом. Она содержит более 102 млн. пар оснований, что составляет 3-3,5% всего материала ДНК клетки человека. Вероятно, она состоит из 700-900 генов.
Примерно в 75% случаев заболевание передается генетически, и лишь 25% случаев заболевания вызываются спорадическими мутациями. Стоит отметить, что СМ характеризуется выраженной генетической гетерогенностью. Сегодня известно около 550 мутаций в разных семьях. Среди выраженных мутаций в гене FBN1: 57% — миссенс мутации, 18% — фреймшифт, 16% — спайс-сайт, 8% — нонсенс мутации.
Зачастую при классической форме СМ характерна мутация в одном из доменов FBN1 (epidermal growth factor EGF-like domain), отвечающих за связывание кальция с фибриллином. Патологические изменения в том же локусе могут вызывать различные клинические проявления — от стертой формы с поражением одной системы организма к классической форме. Клиническое разнообразие СМ обуславливает вовлечение мутаций, локализованных в других генах, например в гене FBN2 или FBN3. Этот факт подтверждается тем, что у части пациентов с клинически выраженным СМ определяется нормальный метаболизм фибриллина, а при генетическом анализе отсутствует мутация в гене FBN1 [Ватутин Н.Т. и др., 2006, Фищенко Я.В., 2006].
Морфология. Молекулярная основа заболевания — нарушение синтеза одного из белков соединительной ткани фибриллина, который придает ей эластичность и обеспечивает сократительную способность. Основные патоморфологические изменения обнаруживаются в соединительной ткани (мезодерме), что в прошлом стало основой названия заболевания, которая сейчас не используется (dystrophia mesodermalis congenita Marfan; dystrophia mesodermalis hypoplastica).Соединительная ткань обладает повышенной способностью к растяжению и менее вынослива к физическим нагрузкам [Ватутин Н.Т. и др., 2006].
Типичные гистологические изменения в средней оболочке сосудов эластического типа, проявляются разрушением эластичного каркаса с некрозом и фрагментацией эластичных волокон, нарушением направленности и расщеплением коллагеновых волокон, дистрофией гладкомышечных клеток, накоплением между волокнистыми структурами мукополисахаридов с последующим формированием небольших кист [Смоленский В.С., 1964, Ватутин Н.Т. и др., 2006].
Классификация. Общепринятой классификации СМ не существует, однако разные авторы пытались это сделать [Лисиченко О.В., 1986].
Классификация синдрома Марфана за Лисиченко:
1. cтертая: слабо выраженные изменения в одной или двух системах.
2. выражена:
— слабо выраженные изменения в трех системах;
— выраженные изменения хотя бы в одной системе; Рис. 2. Снимок черепа — выраженные изменения в двух, трех и более системах. человека, больного долихоцефалией
Степень выраженных изменений:
а) легкая;
б) средняя;
в) тяжелая.
II. Характер течения:
1. Рецидивирующее (прогрессирующее).
2. Стабильное.
III. Генетическая характеристика:
1. Семейная форма (тип наследования).
2. Спорадический случай.
3. Первичная мутация
Клиника. Пациенты с выраженными проявлениями СМ имеют: долихоцефалию; узкий лицевой скелет;высокий рост; низкую массу тела; длинные конечности и паукообразные пальцы; высокое небо; кифосколиоз; воронкообразную грудную клетку, а также поражение органов зрения (эктопию хрусталиков, микросферофакию;
плоскую роговицу, миопию(близорукость)) .
Тяжелая патология сердечно-сосудистой системы.
Различают три вида сердечно-сосудистых проявлений при СМ — врожденные дефекты структуры стенок сосудов эластического типа, особенно в аорте и легочной артерии;
— последствия предыдущих дефектов — аневризмы и разрывы аорты;
— различные пороки развития в сочетании с СМ, например коарктация аорты, гипоплазия аорты, незаращение артериального протока [Смоленский В.С., 1964].
Аневризма аорты при этой патологии возникает с одинаковой частотой у мужчин и женщин, в возрасте 30-40 лет, с преимущественным поражением восходящей част. Имеет мешкообразный вид, с характерным поражением ее ветвей, а также изолированными аневризмами артерий [Зербино Д.Д, 2006] . Поскольку сосудистая патология при синдроме Марфана генерализованная, то поражается эластичная ткань всех сосудов. Аневризмы могут возникать не только в разных отделах аорты, но и в легочной артерии, а также в сонных, лучевых, локтевых, бедренных и других сосудах организма [Ватутин Н.Т. и др., 2006].
Рис. 3. Деформация грудины
(воронкообразная грудная клетка)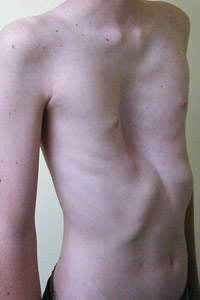 Изменения скелета встречаются у 2 / 3 пациентов, больных синдромом Марфана и включают высокий рост, астеническое телосложение, долихостеномелию, прогнатию, «готическое» небо, деформацию грудины, арахнодактилию, сколиозы, кифосколиозы, нарушение функции суставов, плоскостопие, дисфункцию височно-нижнечелюстного сустава .
Изменения скелета встречаются у 2 / 3 пациентов, больных синдромом Марфана и включают высокий рост, астеническое телосложение, долихостеномелию, прогнатию, «готическое» небо, деформацию грудины, арахнодактилию, сколиозы, кифосколиозы, нарушение функции суставов, плоскостопие, дисфункцию височно-нижнечелюстного сустава .
Офтальмологические проявления диагностируют практически у всех больных СМ, независимо от возраста. Чаще всего это миопия разной степени, плоская роговица, гипоплазия радужки и цилиарной мышцы, эктопия хрусталиков, изменения калибра сосудов сетчатки, косоглазие, дегенерация сетчатки, отслойка сетчатки, катаракта, глаукома. Однако два последних критерии требуют дальнейшей проверки для включения их в категорию малых критериев. Плоская роговица коррелирует с вывихом (подвывихом) хрусталиков [Ватутин Н.Т. и др., 2006, Викторова И.А., 2004, Ольхова О.В., 2010, Mema V., 2010].
Диагностика. По критериям Ghent nosology, 1996 (табл.1) СМ определяется большими и малыми клиническими признаками: в органах опорно-двигательного аппарата; органа зрения; сердечно-сосудистой системы; дыхательной системы; кожи; твердой мозговой оболочки. Для установления диагноза СМ необходимо присутствие по одному крупному критерию в двух системах и одного малого в третьей [Фищенко В. Я., 2007, Moshe Frydman, 2008]. Кроме диагностических критериев, используют фенотипические диагностические тесты СМ, а именно:
— соотношение кисть / рост> 11%;
— отношение размах рук / рост> 1,05;
— длина среднего пальца> 10см;
— отношение длины верхнего сегмента тела к нижнему <0,86;
— индекс Варги <1,5 [Ватутин Н.Т. и др., 2006].
Таблица 1
Диагностические критерии синдрома Марфана
| Диагностические критерии | |
|
| |
Опорно-двигательный аппарат | ≥ 4 из следующих критериев: — килевидная деформация грудной клетки; — изменение соотношения верхний-нижний сегмент возрасту <0,85 или размах рук / рост> 1,05) 1; — плоская стопа; — протрузия вертлюжных впадин; | 2 большие критерии или 1 большой и ≥ 2 из следующих критериев: — гипермобильность суставов; |
Глаза | эктопия хрусталика | ≥ 2 из следующих критериев: |
Сердечно-сосудистая | — дилятация восходящего отдела аорты с участием синусов Вальсальвы; | ≥ 1 из следующих критериев: |
| 1 из следующих критериев: | |
Кожа/мягкие ткани | — атрофические стрии | |
Твердая мозговая оболочка | пояснично-крестцовая эктазия твердой мозговой оболочки (диагностирован на КТ или МРТ) | |
Семейность/наследственность | ≥ 1 из следующих критериев: | |
Дифференциальная диагностика. Поскольку для СМ характерным является поражение костно-мышечной, сердечно-сосудистой систем и органов зрения,то нужно проводить дифференциальную диагностику для того чтобы исключить заболевание на другие синдромы, для которых характерно поражение этих органов и систем:
— Скелетные проявления — синдром Лойса-Дитца, врожденная арахнодактилия с контрактурами (синдром Билса), синдром Стиклера, синдром Клайнфельтера гомоцистинурия, марфаноподобная умственная отсталость;
—
Глазные проявления
— синдром Стиклера, гомоцистинурия (эктопия хрусталика), синдром Элерса-Данло, эктопия хрусталика Вайля-Марчезани, аутосомно-доминантная эктопия хрусталика, аутосомно-рецессивная эктопия хрусталика с/без эктопии радужки;
—
Сердечно-сосудистые проявления
– синдром Лойс-Дитца, синдром Марфана 2 типа, пролапс митрального клапана, МАSS фенотип (семейный пролапс митрального клапана), кистозный медиальный некроз Ердгейма с расслоением восходящей аорты, семейная аневризма аорты, двустворчатый аортальный клапан с расслоением восходящей аорты [ Барашнев, Кадурина, Лисиченко, Umamahesh, Моше Фридман].
клапана, МАSS фенотип (семейный пролапс митрального клапана), кистозный медиальный некроз Ердгейма с расслоением восходящей аорты, семейная аневризма аорты, двустворчатый аортальный клапан с расслоением восходящей аорты [ Барашнев, Кадурина, Лисиченко, Umamahesh, Моше Фридман].
Т. Mizuguchi (и др.) сообщили, что причиной СМ может быть нарушение обмена трансформирующего фактора роста β (ТGFRB2). Бельгийские ученые Барт Лоец и пел. в 2006 году сообщили, что мутации в TGFBR1 является причиной аневризмы аорты и марфаноидного фенотипа (Loeys-Dietz синдром).Хотя расслоения аорты является общим для обоих синдромов (Марфана и Лойс-Дитца), в последнем, эктопии хрусталика является редким симптомом, а у пациентов имеются гипертелоризм (90%), волчья пасть или язычок (90%) и завитки артерий (84%) .
Прогноз. Если не производить лечение болезни Марфана, то в 50% случаев поражения сердца и сосудов приводят к смерти при отсутствии хирургического вмешательства. Наиболее опасной является прогрессирующая дилатация кольца аортального клапана и расслоение аорты. Однако, за последние 19 лет госпитальная летальность при плановых операциях в Национальном институте сердечно-сосудистой хирургии имени Н.Н. Амосова в Украине составила 0%, при 164 прооперированных. Своевременная кардиохирургическая коррекция сердечно-сосудистых проявлений и осложнений синдрома Марфана позволила увеличить продолжительность и улучшить качество жизни многих пациентов с СМ [Хорошковатого О.В., 2010].
Лечение. В лечении СМ используют медикаментозное и хирургическое лечение, одинаково улучшают продолжительность жизни больных до 60-70 лет. Медикаментозная терапия должна быть продолжена, и после операции. Среди медикаментов применяют β-блокаторы, которые снижают скорость расширения аорты и могут увеоичить продолжительность жизни пациентов. Целью лечения должен быть строгий контроль над артериальным давлением (систолическое давление до 120 мм рт.ст. и 110 мм рт.ст. для пациентов с расслоением аорты). Большинство центров мира используют именно β-блокаторы (бисопролол). Блокаторы рецепторов ангиотензина II являются потенциально полезными, поскольку они приводят к антагонизму ТGF-β. Клинические испытания, которые проводятся сейчас, показывают хорошую эффективность лозартана для профилактики аневризмы аорты у больных СМ (ESC Guidelines, 2010).
Сегодня существует три основных подхода к хирургическому лечению сердечно-сосудистых осложнений у больных СМ:
—
во-первых
— протезирование восходящей аорты и аортального клапана с использованием кондуита, который содержит клапан (операция Bentall de Bono);
—
во-вторых
— отдельное протезирование аортального клапана и восходящей аорты в случае, если синусы Вальсальвы не изменены, но наблюдаются выраженные деструктивные изменения в створках аортального клапана;
—
в-третьих
— выполнение клапано-сохраняющих операций в случае интактности аортального клапана или умеренно выраженных морфологических его изменений.
Литература:
1. Барашнев Ю.И., Казанцева Л.З., Семячкина А.Н., Бухны Л.Ф. (1983) Дифференциальный диагноз болезни Марфана и некоторых сходных с ним синдромов (синдром Билса и Стиклера) у детей // Вопросы охраны материнства и детства, № 4. С. 41-46.
2. Белоконь Н.А., Кубергер М.Б. (1987) Болезни сердца и сосудов у детей: Руководство для врачей. В 2 т. Т. 2. – М.: Медицина, – 480 с.
3. Богомолов Л. И., Фисанович Т.И., Карлова Т.Ф. (1982) Поражение периферических артерий при синдроме Марфана // Клиническая хирургия, № 7. С. 66.
4. Ватутин Н.Т., Склянная Е. В., Кетинг Е.В. (2006) Синдром Марфана // Кардиология, № 1. С. 92-98.
5. Викторова И.А., Нечаева Г.И.(2004) Синдром Марфана в практике терапевта и семейного врача: диагностика, тактика ведения, лечение, беременность и роды // Русский медицинский журнал, №2.
6. Демин А.А., Антонов О. С., Семенова Л. А., Сентякова Т.Н., Карчова Т. Ю., Муранова Г. В., Рыскинд В.А., Кузнецов В.А. (1985) Синдром Марфана: полиморфизм клинических проявлений // Терапевтический архив, т. 57, № 4, С. 133-135.
7. Дмитриев В. И., Марченко А. М., Марин А. И. (1981) Особенности клиники синдрома Марфана в юношеском и зрелом возрасте // Врачебное дело, №7, С. 78-80.
8. Зербіно Д. Д. (2006) Патологія аорти: класифікація, хвороби і синдроми, проблеми етіології // Медицина транспорту України, № 2, С. 6-14.
9. Кадурина Т. И., Горбунова В. Н.(2009) Дисплазия соединительной ткани. Руководство для врачей. – СПб.: Элби – СПб. – 704 с.
10. Контридзе В. С., Кванталиани И. Г. (1980) Три случая трансплантации склеры при синдроме Марфана // Вестник офтальмологии, №4. С. 68-69.
11. Кравченко І.М., Сітар Л.Л., Федонюк Л.Я., Захарова В.П.(2007) Аневризми висхідної аорти та аортальна недостатність при синдромі Марфана: проблеми хірургічного лікування та морфології // клінічна анатомія та оперативна хірургія, №4, С. 58-61.
12. Кузьмінський А.П., Малярська Н.В., Опалинський Ю.А. (2008) Гострі порушення мозкового кровообігу і синдром Марфана // Буковинський медичний вісник, № 3. С. 123 – 125.
13. Лазовскис И. Р.(1981) Справочник клинических симптомов и синдромов. 2-е изд., перераб. и доп. – М.: Медицина, — 512с.
14. Лайбер Б., Ольбрих. Г. (1974) Клинические синдромы. М.: Медицина. – 477с.
15. Леванюк В.Ф., Тидир А. А. (1983) Диагностическая ценность индекса телосложения Варги при синдроме Марфана // Клиническая медицина, т.61, №9. С. 131-134.
16. Лисиченко О. В.(1986) Синдром Марфана. – Новосибирск: Наука, – 164с.
17. Ольхова О.В. (2010) Родинна форма синдрому Марфана: варіанти вад очей // Мистецтво лікування, №6 (72), с.110 – 112.
18. Оспанова Л.С., Вятчинин Н.Г., Турлубеков К.К.(1986) Синдром Марфана // Здравоохранение Казахстана, №10. С. 70-71.
19. Смоленский В.С. (1964) Болезни аорты . – М.: Медицина. – 283с.
20. Фіщенко В. Я., Фіщенко Я. В. (2007) Семіотика синдрому Марфана. Укр. мед. альманах, 10(2): 172–175 с.
Источник