Прогрессирует ли синдром арнольда киари

Общая информация
Синдром Арнольда-Киари представляет собой набор признаков и симптомов, вызванных редкой мальформацией (отклонение от нормального развития, аномалия) задней черепной ямки; у пострадавших эта структура развита слабо, поэтому мозжечок выходит (выступает) из своего естественного участка через затылочное отверстие, расположенное у основания черепа.
Есть четыре различных типа синдрома Арнольда-Киари; особенность, отличающая один тип от другого, является степень выпячивания, следовательно, доля вовлеченного материала мозжечка. Тип I является наименее тяжелым (иногда остается бессимптомным на протяжении всей жизни), тогда как IV тип наиболее тяжелый; однако уже со второго типа качество жизни больного ставится под угрозу.
Симптомы, характеризующие аномалии Арнольда-Киари многочисленны и варьируются от головных болей до слабости мышц и проч.
На сегодняшний день не существует лекарств, позволяющих устранить порок развития мозжечка, однако существуют способы лечения, позволяющие частично смягчить симптомы.
Что такое синдром Арнольда-Киари?
Синдром Арнольда-Киари, или мальформация Арнольда-Киари — структурное изменение мозжечка, характеризующееся его смещением вниз, именно в направлении позвоночного канала и затылочного отверстия, базальные части полушария мозжечка.
Простыми словами, это грыжа мозжечка, при которой часть мозжечка выступает из затылочного отверстия, проникая в позвоночный канал.
Синдром Арнольда-Киари получил свое название от двух врачей, которые впервые описали его, Арнольда Джулиуса и Ганса Киари.
Причины и факторы риска
Исследователи полагают, что синдром Арнольда-Киари может иметь наследственное происхождение, так как обнаруживалась среди членов одной семьи. Тем не менее, генетические условия, вызывающие заболевание (т.е., какие и сколько генов участвуют) и тип передачи еще предстоит выяснить.
Исходя из серьезности выпячивания и момента жизни, в котором он возникает, заболевание можно разделить на 4 различных типа, идентифицированных первыми четырьмя римскими числами (I, II, III и IV).
Первые два типа по сравнению со вторыми более распространены и менее серьезны; Тип III и тип IV, на самом деле, очень редки и несовместимы с жизнью.
— Мальформация I типа.
Первая степень синдрома протекает бессимптомно (т.е. без явных симптомов), по крайней мере, до конца детства или юности.
Причина его возникновения кроется в уменьшенном черепном пространстве: в таких условиях часть мозжечка (именно миндалина(и), расположенная(ые) с нижней стороны), из-за недостатка места, вынуждена проникать в затылочное отверстие и входить в позвоночный канал.
Примечание: у некоторых взрослых людей с синдром Арнольда-Киари 1 типа все в порядке и они ведут совершенно нормальную жизнь. Это связано с тем, что аномалия мозжечка не настолько серьезна, чтобы вызывать симптомы или нарушения. Поэтому очень часто эти субъекты игнорируют свое состояние или узнают о нем по чистой случайности.
— Мальформация II типа.
2 тип мальформации Арнольд-Киари является врожденным заболеванием, которое присутствует с рождения ребенка, и всегда протекает симптоматически.
По сравнению с 1 степенью он характеризуется большим выпячиванием черепной ямки, при котором помимо миндалин мозжечка также выпячивает часть мозжечка (называемая червь мозжечка) и венозный сосуд.
Почти всегда мальформация Арнольд-Киари II типа ассоциируется с особой формой расщелины позвоночника, называемой миеломенингоцеле.
Среди различных последствий этой аномалии выделают: блокирование потока ликвора (спинномозговой жидкости) через затылочное отверстие (что приводит к состоянию, называемому гидроцефалией) и прерывание нервных сигналов.
Первоначально термин Арнольд-Киари относился только ко 2 типу заболевания. Теперь, её обычно используют для всех форм болезни.
— Мальформация III типа.
Присутствующий с рождения, III тип порока вызывает серьезные неврологические проблемы, настолько, что часто несовместимы с жизнью. В этих случаях на самом деле наблюдается выпячивание мозжечка, и по этой причине говорится о затылочном энцефалоцеле.
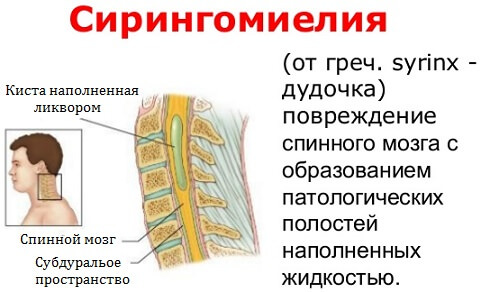
Обычно III тип состояния характеризуется гидроцефалией и сирингомиелией; последний представляет собой особое состояние, характеризующееся наличием одной или нескольких кист в позвоночном канале.
— Мальформация IV типа.
Мальформация Арнольда-Киари IV типа характеризуется отсутствием развития части мозжечка (недоразвитие мозжечка).
Аномалия врожденная и абсолютно несовместима с жизнью.
Связанные расстройства
Врачи и ученые отметили, что следующие заболевания являются частыми среди людей с мальформацией Киари:
- гидроцефалия;
- сирингомиелия;
- сколиоз;
- синдром Марфана;
- синдром Элерса-Данлоса.
Эпидемиология
Точная частота пороков развития неизвестна; это связано с тем, что у некоторых даже взрослых людей с I типом мальформации Арнольда-Киари никаких симптомов нет, и они кажутся совершенно нормальными (поэтому болезнь недиагностируется).
Несколько достоверных эпидемиологических исследований сообщают, что:
- I тип симптоматичен у 1 из 100 детей;
- II тип особенно широко распространен в популяциях кельтского происхождения;
- женщины страдают в 3 раза чаще, чем мужчины.
Симптомы и осложнения

4 типа заболевания имеют разные симптомы и признаки.
Ниже приводится таблица с точным описанием симптомов, которые характеризуют I, II и III типы синдрома.
Для IV типа невозможно проследить симптоматику, так как это состояние неизбежно и внезапно приводит к гибели плода.
| Мальформация I типа | Мальформация II типа | Мальформация III типа |
Когда у больного 1 стадия, симптомы следующие:
| Синдром Арнольда-Киари II типа характеризуется теми же симптомами, что и тип I, с той разницей, что они имеют более выраженную интенсивность и присутствуют всегда. Кроме того, если он сопровождается миеломенингоцеле (см. ниже), II тип состояния также вызывает:
| Люди с мальформацией III типа страдают от серьезных неврологических проблем (часто несовместимых с нормальной жизнью), гидроцефалии и сирингомиелии. Последний, характеризуется образованием одной или нескольких кист внутри спинного мозга, и может вызывать:
|
Расщелина позвоночника (миеломенингоцеле).
Расщелина позвоночника — врожденный порок развития позвоночника, из-за которого менинги, а иногда и спинной мозг выходят из своего места (обычно они ограничены позвонками). Миеломенингоцеле является наиболее тяжелой формой расщелины позвоночника: у пораженных выпячивают менинги и спинной мозг из позвоночной камеры и образуют мешок на уровне спины. Эта сумка, хотя и защищена слоем кожи, подвержена внешним воздействиям и постоянно подвергается риску серьезных, а в некоторых случаях даже смертельных инфекций.
Синдром Арнольда-Киари типа II, III и IV видны уже в пренатальном возрасте (т.е., когда пораженный ребенок все еще находится в материнской утробе) при ультразвуковом исследовании.
Что касается I типа, желательно обратиться к врачу, как только появятся типичные симптомы, о которых говорилось выше. Важно также проходить своевременные обследования, так как в результате последних могут возникнуть другие сопутствующие нарушения.
Осложнения
Осложнения синдром Арнольда-Киари связаны с ухудшением выпячивания мозжечка или патологическими состояниями, связанными, следовательно, с гидроцефалией, миеломенингоцеле, сирингомиелией и проч.
Ухудшение выпячивания (протрузии), обусловленное повышенным давлением черепа на мозжечок, очевидно, предполагает обострение симптомов.
Диагностика
Диагностические тесты, которые позволяют установить степень протрузии мозжечка через затылочное отверстие (таким образом, устанавливая тип мальформации Арнольда-Киари):
- Магнитно-резонансная томография (МРТ). Благодаря формированию магнитных полей, он позволяет получить детальное изображение мозжечка и позвоночного канала, не подвергая пациента вредному ионизирующему излучению.
- Компьютерная томография (КТ) дает четкие изображения внутренних органов, в том числе мозжечка и спинного мозга. Во время его выполнения субъект подвергается минимальному воздействию вредного ионизирующего излучения.
КТ и МРТ, которым предшествует точное физическое обследование, имеют основополагающее значение для выявления любых патологий, связанных с синдромом Арнольда-Киари.
Таблица. Как и когда диагностируется аномалия Арнольда-Киари развития заболевания.
| Тип порока развития | Когда и как это можно диагностировать? |
| I | В позднем детстве или позднем подростковом возрасте посредством объективных обследований с последующим КТ и/МРТ. |
| II | В дородовом возрасте при УЗИ. При рождении и в раннем детстве, посредством объективных обследований, компьютерной томографией и/или МРТ. |
| III | В дородовом возрасте при УЗИ. После рождения и в раннем детстве, посредством объективных обследований, компьютерной томографией и/или МРТ. |
| IV | В дородовом возрасте при УЗИ. |
Лечение
Синдром Арнольда-Киари неизлечим. Однако существуют как фармакологические, так и хирургические методы терапии, позволяющие частично смягчить признаки заболевания.
— Медикаментозная терапия.
Пациенты с мальформацией Арнольда-Киари I типа, страдающий от головной боли и боли в шее и/или лице, могут принимать обезболивающие препараты.
Выбор наиболее подходящих лекарств для конкретного случая остается за лечащим врачом.
— Хирургическое лечение.
Цель хирургического лечения состоит в том, чтобы уменьшить давление, оказываемое черепом, чтобы предотвратить повреждение мозжечка и спинного мозга.
Для достижения этой цели есть несколько процедур, таких как:
- Декомпрессия задней черепной ямки, во время которой хирург удаляет часть задней части затылочной кости.
- Декомпрессия спинного мозга с помощью ламинэктомии (декомпрессионная ламинэктомия). Во время его выполнения хирург удаляет пластинку второго и третьего шейного позвонка. Пластинка — это позвоночная часть, отделяющая отверстие, через которое проходит спинной мозг..
Примечание: иногда декомпрессия задней ямки и декомпрессивная ламинэктомия выполняются одновременно. - Декомпрессионный разрез твердой мозговой оболочки. При разрезе твердой мозговой оболочки или наружного менинга пространство, доступное мозжечку, увеличивается, а давление на его повреждение уменьшается. Чтобы покрыть и защитить трещину, созданную разрезом, хирург пришивает на нее кусок искусственной ткани (или взятый из другой части тела).
- Хирургическое шунтирование (создание дополнительного пути в обход пораженного участка). Это, по сути, дренажная система, состоящая из гибкой трубки, позволяющей удалять спинномозговую жидкость, в случае гидроцефалии, или опорожнять цисту(ы), в случае сирингомиелии. Не исключено, что пациенты с гидроцефалией будут вынуждены проходить хирургическим шунтом всю жизнь.
— Осложнения хирургического вмешательства.
Риски, связанные с хирургией, различны. Фактически возможно появление:
- кровотечений;
- повреждение структур головного мозга и/или спинного мозга;
- инфекционный менингит;
- проблемы с заживлением ран;
- необычные скопления жидкости вокруг мозжечка.
Помните, что любое повреждение головного или спинного мозга, произошедшее во время операции, непоправимо. Поэтому, прежде чем подвергнуться больного вмешательству любого типа, лечащий врач выявит любые риски и осложнения необходимой процедуры.
Прогноз
Синдром Арнольда-Киари типа II, III и IV никогда не имеют положительного прогноза, поскольку, помимо того, что неизлечимы, могут вызывать серьезные неврологические нарушения или даже быть несовместимыми с жизнью.
Прогноз для пациентов с I типом часто неизвестен. Многие люди с этим заболеванием не имеют никаких симптомов, и невозможно предсказать, будут ли симптомы развиваться в будущем. Другие люди с мальформацией Арнольда-Киари могут испытывать головокружение, мышечную слабость, онемение, проблемы со зрением, головную боль или проблемы с равновесием и координацией. У этих людей не всегда возможно предсказать, будут ли симптомы ухудшаться с течением времени.
Людям с пороком 1 типа важно регулярно проходить медицинские обследования, чтобы быть под наблюдением врача при появлении любых новых симптомов.
Источник
Общие сведения
Синдром Арнольда-Киари является пороком развития мозжечка – отдела головного мозга, отвечающего за координацию, мышечный тонус и равновесие. Патологии присвоен код по МКБ-10 Q07.0 и она представляет собой опущение миндалин мозжечка вниз на уровень первого, а порой второго шейного позвонка (ниже черты Чемберлена) и блокирует нормальный ток спинномозговой жидкости.
Заболевание чаще всего сочетается с микрогирией, сдавливанием заднего отдела мозга, стенозом водопровода мозга, базилярной импрессией, инвагинацией, недоразвитием четверохолмия и другими мальформациями нервной системы. Синдром чаще всего встречается у особ в возрасте 12-71 год и не превышает 000,9%.

Локализация и строение мозжечка
Патогенез
В основе патофизиологии обычно лежит несоответствие размеров задней черепной ямки и имеющихся в ней структур нервной системы, а также:
- развитие аномалий тел шейных позвонков, включая их расщепление чаще всего первого (данный механизм развития встречается в 5% случаев), ассимиляцию атланта — сращивание шейного позвонка с затылочной костью;
- смещение структур мозжечка в период бурного роста мозга при медленно растущих костях черепа;
- гидроцефалия – избыточное скопление цереброспинальной жидкости;
- сирингомиелия – анормальный процесс развития полостей в спинном мозге;
- миеломенингоцеле – врожденный дефект развития нервной трубки;
- различные врожденные заболевания, в том числе платибазия, аномалия Денди-Уокера.
Классификация
В зависимости от клинической картины и степени развития анатомических аномалий синдром Арнольда-Киари бывает четырех типов.
Аномалия Арнольда Киари 1 типа – это классический вариант порока развития мозжечка
Синдром Арнольда-Киари 1 типа проявляется в виде проникновения миндалин мозжечка в полость позвоночного канала, вызывающего гидромиелию и опущение структур задней черепной ямки ниже большого затылочного отверстия на 3-5 мм и более, причем нет никаких других мальформаций нервной системы. Средняя продолжительность жизни обычно не превышает 25-40 лет.
Порок мозжечка 2 типа
Представляет собой опущение в полость позвоночного канала различных структур мозжечка и тканей ствола, при этом данная нейропозвоночная мальформация сочетается с миеломенингоцеле (врожденной спинномозговой грыжей) и гидроцефалией. Манифестация происходит практические сразу после рождения.
Мальформация Арнольда Киари 3 типа
Мальформация отличается наличием затылочного энцефалоцеле и различных признаков аномалии второго типа. Обычно не совместима с жизнью.
Четвертый тип Синдром Арнольда — Киари
По своей сути это аплазия либо гипоплазия всех или отдельных структур мозжечка, то есть бывает тотальной и субтотальной. Первый вариант встречается достаточно редко и сочетается с прочими тяжёлыми аномалиями и заболеваниями нервной системы, включая анэнцефалию, амиелию. При субтотальной агенезии наблюдаются пороки развития других участков головного мозга, например агенезия моста, отсутствие четвёртого желудочка и пр.
Гипоплазия мозжечка встречается в форме уменьшения всего мозжечка или охватывает отдельные части, при этом сохраняются нормальные структуры без утраты функций. Встречается одно- и двусторонняя, лобарная, лобулярная и интракортикальная гипоплазия. Изменения конфигурации листков мозжечка обычно представлено в виде аллогирии, полигирии или агирии.
Кроме того, некоторые авторы выделяют два дополнительных типа:
- тип «0» — заболевание имеет сходную клиническую картину с синдромом Арнольда Киари, но без анатомических изменений опущения миндалин или гипоплазии мозжечка;
- тип «1,5» — у больных выявляется опущение не только миндалин мозжечка в затылочное отверстие, но и ствола головного мозга.
Причины
Помимо роли наследственного и генетического фактора существует несколько теорий возникновения пороков мозжечка. Традиционная теория говорит, что опущение миндалин вызвано натяжением струны спинного мозга в результате напряжения концевой нити при развитии той или иной мальформации. Исключением становится болезнь Киари 1 типа, ведь единственным нарушением в этом случае становится опущение миндалин и оно может быть спровоцировано:
- гидродинамическими явлениями — нарушением циркуляции спинно-мозговых жидкостей;
- черепно-мозговыми и родовыми травмами;
- мальформацией — маленькие размеры и ограниченность затылочного отверстия могут приводить к опущению миндалин в просвет позвоночного канала;
- анормально натянутой связкой — так называемой концевой нитью (по теории доктора М.Б. Ройо Сальвадора — Filum System).
Кроме того, ученые выделяют ряд факторов, которые могут повысить риск развития аномалии Арнольда-Киари первого типа:
- генетическая предрасположенность – наличие патологии у родителей и более дальних предков, хотя хромосомных аномалий до сих пор не выявлено;
- травмы, особенно падения, могут вызвать компрессию и усиление натяжения концевой нити и привести к опущению миндалин мозжечка;
- неправильное поведение и вредные привычки женщины в период вынашивания младенца — злоупотребление и хаотичный прием медикаментов, курение, употребление алкоголя, а также перенесенные вирусные заболевания.
Симптомы
Симптоматика при различных типах синдрома Арнольда Киари может существенно отличаться – все зависит от степени натяжения и компрессии нервных структур затылочного отдела, но чаще всего у больных наблюдаются:
- головные боли;
- прогрессирующее увеличение размеров окружности головы;
- периодические боли в различных отделах позвоночника;
- парез и боль в различных областях конечностей;
- нарушения зрения и чувствительности, включая дизестезии и парестезии;
- шумы в ушах;
- паралич лицевого, глазодвигательного нерва;
- дрожь;
- приступы головокружения;
- бессонница;
- рвота;
- обмороки;
- нистагм;
- апноэ;
- нарушение работы сфинктеров, мышц языка, глотки, онемение и различные проявления мышечной слабости;
- нарушения способности глотания (дисфагия);
- нарушения памяти;
- несогласованность движений (атаксия) и как следствие — неуклюжая походка, причем нарушения наиболее ярко выражены на этапе формирования;
- сколиоз;
- трудности с удержанием позиций тела и равновесия, а также координации;
- затруднения при желании выразить мысль и подобрать слова.
Проявления синдрома Арнольда-Киари хронические, имеют тенденцию увеличивать интенсивность, что с каждым разом существенно ухудшает состояние больного и ограничивает его привычный образ жизни.
Самое опасное, что аномалия Арнольда-Киари может привести к внезапной смерти, ведь спинно-мозговые центры отвечают за сердечно-дыхательные функции, а давление миндалин мозжечка на них может спровоцировать остановку дыхания — апноэ, которое станет причиной летального исхода.
Симптомы, которые вызывает аномалия Арнольда Киари 1 степени
Патология 1 степени обычно выявляется случайно во время проведения МРТ, ведь больные помимо эпизодов апноэ и обмороков могут испытывать только:
- боль в шейно-затылочном отделе;
- снижение чувствительности.
Анализы и диагностика
При первых признаках необходимо пройти неврологический осмотр и провести оценку выраженности клинико-функциональных нарушений. Для подтверждения диагноза чаще всего применяются:
- нейровизуализационной методики, наиболее предпочтительно МРТ;
- электроэнцефалография;
- рентгенография черепа;
- миелография, которая позволяет выявить дефекты верхнего шейного отдела спинного мозга, ствола мозга, а также локализацию мозжечка ниже черты Чемберлена в области затылочного отверстия.

Результаты МРТ при аномалии Арнольда Киари с сирингомиелической кистой и в норме
Лечение
Тактика лечения при различных типах пороков развития мозжечка Арнольда-Киари обычно является консервативной либо продумывается нейрохирургом и представляет собой декомпрессию, наложение шунта (при выраженной гидроцефалии) или краниотомию затылочного отверстия.
Однако, благодаря докторской диссертации доктора М.Б. Ройо Сальвадора была разработана новаторская техника этиологического лечения — Filum System, которая направлена на устранение причины заболевания и патологического механизма натяжения путем хирургического минимально инвазивного рассечения концевой нити. Преимуществом методики является возможность остановить болезнь Арнольда Киари при минимальных рисках (смертность – 0%), главное выявить мальформацию и провести операцию как можно раньше. Она обычно занимает не более 45 минут и позволяет добиться симптоматического улучшения состояния, а в отдельных случаях даже поднятия миндалин мозжечка. Несмотря на короткий восстановительный период, методика имеет ряд недостатков:
- после операции остается небольшой шов;
- может возникать субъективное ощущение снижения силы конечностей;
- улучшение мозгового кровообращения вначале постоперативного периода может вызвать перепады настроения.
Но это незначительные неудобства по сравнению с минусами затылочной краниотомии, у которой:
- смертность 1-12%;
- причина заболевания не устраняется, поэтому улучшения сохраняются непродолжительный период;
- последствия оперативного вмешательства могут быть очень серьёзными и непредсказуемыми, включая отек мозга, дальнейшее опущение миндалин мозжечка, усугубление неврологической симптоматики, гемодинамические нарушения, гидроцефалию, пневмоэнцефалию, внутримозговые кровоизлияния, тетрапарез, неврологический дефицит и т.д.
Доктора
Лекарства
Медикаментозная терапия обычно симптоматическая, чаще всего пациентам назначают НПВС, миорелаксанты и анальгетики. Например, Прегабалин, Карбамазепин, Спазмалгон, Палексия и пр.
Проблемой становится неэффективность противовоспалительных и обезболивающих препаратов при купировании приступов головной боли или других симптомах.
Процедуры и операции
- лечебная гимнастика (кинезотерапия);
- избирательный точечный массаж;
- иглорефлексотерапия и электроакупунктура при наличии мышечной спастичности и болевых синдромов;
- занятия с логопедом (при необходимости коррекции нарушений речи);
- психокоррекционная работа психолога при наличии эмоциональных, дисфорических или депрессивных расстройств.
У детей
Чаще всего отмечается блокада внутри желудочковой системы и представляет собой несообщающуюся гидроцефалию. Это обычно вызвано сужением сильвиева водопровода либо недоразвитием отверстий Мажанди и Люшки. Как генерализованное нарушение развития нервной системы, оно может сочетаться с микро- или макрогирией, порэнцефалией, агенезией мозолистого тела, слиянием полушарий мозга, агенезией червя мозжечка, позвоночных расщелин, менингоцеле, энцефалоцеле, сирингомиелией, гидромиелией. Причиной сообщающейся гидроцефалии может быть процесс образования спаек в субарахноидальной области в основания мозга, перинатальные субарахноидальные или внутрижелудочковые кровоизлияния.
Синдром у детей может возникать в результате травматических повреждений клиновидно-затылочной и клиновидно-решетчатой области при родовых травмах. Клинические проявления сводятся к нарушению кормления (рефлюксу и аспирации), эпизодам апноэ, стридору, нарушениям краниальной иннервации, судорожным приступам устойчивым к противосудорожным препаратам.
Синдром Арнольда Киари у плода
Патология Киари является врожденным остеоневропатией и формируется на этапе закладки и развития нервной трубки у плода, асинхронным формированием нервного ствола и костей черепной ямки.
Морфологические проявления данной патологии можно выявить во время проведения ультразвукового исследования, предопределить дальнейший прогноз. Чаще всего выявление патологии становится причиной для прерывания беременности.
Список источников
- Петровский Б.В. Большая медицинская энциклопедия — М.:: Советская энциклопедия, 1981. — Т. XV (Меланома-Мудров). — С. 576.
- Гусев Е. И., Коновалов А. Н., Скворцова В. И. Неврология и нейрохирургия. — М.: ГЭОТАР-Медиа, 2010.— 624 с.
Источник